单、 双及双混氮杂卟啉的结构和电子吸收光谱
2020-02-19曹洪玉马子辉张文琼李如玉郑学仿
曹洪玉, 马子辉, 张文琼, 唐 乾, 李如玉, 郑学仿,
(1. 大连大学生命科学与技术学院, 2. 环境与化学工程学院, 大连 116600)
卟啉具有特殊的吸收光谱, 能够有效传递电子且化学稳定性较高[1,2], 可用于染料敏化太阳能电池[3,4]、 光学材料[5]及光动力学治疗光敏剂[6]等方面的研究. 源于叶绿素仿生的卟啉类材料具有明显的优势, 为了能够更高效地利用太阳能, 研究通过修饰卟啉[7], 使其在紫外-可见光区具有更强的吸光强度和更宽的吸收谱带, 提高对光的利用率[8,9], 可获得更理想高效的绿色卟啉类材料. Kelly和Snell[10]采用血卟啉首次开创了光动力学疗法(PDT)治疗膀胱癌患者, 现已成为临床上肿瘤治疗常用的方法之一[6,11], PDT中的核心材料是光敏剂, 其中传统卟啉类敏化剂有血卟啉衍生物[12]和酞菁类衍生物[13]等. 近年来, 人们设计合成了不同卟啉及其金属配合物以期获得更优良的光敏剂, 如Bryant等[14]通过制备铱卟啉络合物使金属卟啉络合物在水中溶解性与抗癌能力等方面得到提升, 但这类卟啉化合物存在一定的缺陷, 如可见光区吸光能力弱(600~800 nm)等.
对卟啉结构的修饰包括降低卟啉的对称性和增大共轭程度等方法[7]. 修饰卟啉结构可改变卟啉化合物性质(如光能吸收、 电子传递等), 以适应其在不同研究领域中的应用. Vogel等[15]首次提出了卟啉异构体, 并合成第一个卟啉的同分异构体Porphycene, 又陆续合成了Corrphycene[16], Hemiporphycene[17], Isoporphycene[18]等化合物, 与原卟啉不同之处在于其吡咯环的连接方式发生改变, 但仍保持了18π电子共轭的芳香性体系. 1994年, Furuta等[19]和Chmielewski等[20]分别合成卟啉真正意义上的同分异构体单氮杂卟啉(NCP), 自此NCP类化合物的结构和光谱性质广受关注; 2011年, Fujino等[21]又报道了一种新的单氮杂卟啉异构体(Neo-CP), 研究发现这2类氮杂卟啉与卟啉具有相似的性质和光谱, 易与金属形成络合物. 随后双氮杂卟啉(DNCPs)[22,23]、 双混杂卟啉(Neo-C-NCPs)[24]和多氮杂卟啉陆续被合成. 上述氮杂卟啉是卟啉的同分异构体, 保留了卟啉18π电子共轭大环结构, 但在电子结构和Q带光吸收性质方面差别较大, 其可见光区吸光能力增强, 有希望应用于PDT光敏材料治疗肿瘤疾病. 目前, 氮杂卟啉合成难度较大, 产率较低, 其光谱性质表征成本较高, 因此氮杂卟啉结构、 能级和吸收光谱的变化及不同溶剂对其理化性质的影响及规律尚缺乏系统研究和筛选方法.
本文采用密度泛函理论(DFT)讨论了单氮杂卟啉、 双氮杂卟啉及双混氮杂卟啉等13种卟啉分子在几何结构及分子轨道能级上的差异. 采用含时密度泛函理论(TD-DFT)研究了在真空及水、 氯仿和苯3种不同极性溶剂对各种氮杂卟啉的电子结构和吸收光谱的影响, 为筛选和合成新的氮杂卟啉类光敏剂材料提供理论依据.
1 计算方法
研究了FBP、 3个单氮杂卟啉、 3个双氮杂卟啉和6个双混杂卟啉等13种卟啉, 化合物命名由N原子取代位置数字和卟啉类型组成, 后面的数字和H表示氢迁移至卟啉外侧的位置. 所有结构的计算均采用Gaussian 09[25]程序包完成. 真空条件下, 几何构型均采用B3LYP/6-31+G(d)基组优化, 采用积分极化连续模型(IEF-PCM)模型[26]分别在水、 苯和氯仿3种溶剂中优化分子的基态几何构型, 方法和基组为B3LYP/6-31+G(d); 频率分析无虚频表明优化的构型稳定, 在相同的基组水平上采用TD-DFT理论[27,28]在真空和3种不同极性溶剂中计算卟啉分子的电子光谱, 激发态均采用B3LYP/6-31+G(d)基组计算, 激发态Nstates设置为120. 利用Multiwfn波函数[29,30]软件分析13种卟啉电子转移情况.
2 结果与讨论
2.1 卟啉的分子构型
采用Gaussian 09程序包, 用DFT/B3LYP/6-31+G(d)优化卟啉异构体分子结构(图 1). 卟啉和单氮杂卟啉(FBP和N/Neo-CPs)的结构数据与实验值[19,31,32]基本吻合, 其中2-NCP-2H的键长最大偏差为0.0124 nm(Cα—Cβ); 卟啉和单氮杂卟啉(FBP和N/Neo-CPs)的几何参数与Liu等[33]以及其它计算[34,35]结果基本一致, 其中最大偏差为0.0016 nm(FBP, N-Cα)(图S1, 见本文支持信息). 可见, 采用的B3LYP/6-31+G(d)对卟啉进行优化的方法是合理的.

Fig.1 Molecular structures of porphyrin and its isomers
结果表明, 氮杂卟啉大环键长均一化程度降低, 环外C—C和N—C键键长略有减小, N/C交换的吡咯环上相关键长减小最显著. 单氮杂卟啉Neo-CPs或N-CPs在几何结构上都有变化, 其中2-NCP-2H变化最小, 键长差最大为0.0041 nm, 2-NCP-2H的大环保持平面结构(图S2, 见本文支持信息). 双氮杂卟啉DNCPs和Neo-C-NCPs中, 有2个吡咯环上碳氮交换, 环内氢原子增加, 导致其卟啉的平面结构扭曲. Neo-C-NCPs分子中结构变化较小的是2,18-DNCP-2H, 与FBP相应键的键长差最大为0.0090 nm, 由于环中心空间斥力较大, 吡咯环与整个分子大环平面形成倾斜角, 其中吡咯环Ⅲ与平面形成最大倾斜角(23.55°). 2,18-DNCP-2H和FBP键长差为0.0069 nm(Nβ—Cβ), 环中心原子间间距差最大为0.0133 nm. 在单氮杂卟啉、 双氮杂卟啉和双混氮杂卟啉中, 由于N/C原子交换导致其卟啉环平面的扭曲, 键长均一化程度下降及原子间距增大, 且平面扭曲按单氮杂卟啉、 双混氮杂卟啉和双氮杂卟啉的顺序增大.
2.2 电荷密度和偶极矩
研究分子中原子的电荷密度, 对预测分子的反应类型和发生反应时电子转移具有重要意义. 在FBP上4个N原子、 4个Cm原子和Cβ原子均带负电荷, Cα原子均带正电荷(图S3, 见本文支持信息). 由于吡咯环上N原子位置的改变以及N, C原子的电负性不同, N/Neo-CPs分子中原子电荷发生显著变化, 其中变化最大的是2-NCP-2H分子, 吡咯环Ⅰ(发生N/C交换)上与氮原子相邻的环内Cα原子的电荷为-0.881 e, 在FBP中相同位置C原子带的电荷为0.403 e, 该现象也发生在其它N/Neo-CPs, DNCPs和Neo-C-NCPs分子上.
在2,18-DNCP分子中, 由于发生N/C交换, Ⅰ, Ⅱ两环上Cα原子与N原子直接相连, 负电荷转移至Cm原子(-1.274 e), 远大于其它Cm原子(-0.369~-0.686 e)(图S3). 在Neo-C-NCPs分子中, 发生N/C交换的吡咯环Cα原子上的电荷分布也存在相同现象, 其中6,17-Neo-C-NCP的2个Cα原子的电荷分别为 -1.025和-0.850 e, 其绝对值明显远大于FBP分子中的相同位置Cα原子的电荷数值. 以上结果表明N原子的位置对氮杂卟啉的电荷分布影响较大, 尤其是大环内N/C交换吡咯环的Cα原子.
FBP对称性较高, 偶极矩为0, N/C原子交换后分子对称性被破坏, 偶极矩发生较大的变化(表1). 溶剂的极化作用对卟啉分子化合物的偶极矩影响也较大, 如2,18-DNCP在苯溶剂中的偶极矩为35.86×10-30C·m, 在水溶剂中偶极矩增至48.21×10-30C·m; 真空条件下偶极矩越大的分子, 受溶剂极性影响越大, 如2,18-DNCP在苯和水溶剂中的偶极矩差为12.35×10-30C·m, 而2,12-DNCP在苯和水溶剂中的偶极矩差值仅为0.133×10-30C·m.

Table 1 Dipole moments(μ) of porphyrin isomers in different polar solvents
2.3 电子结构
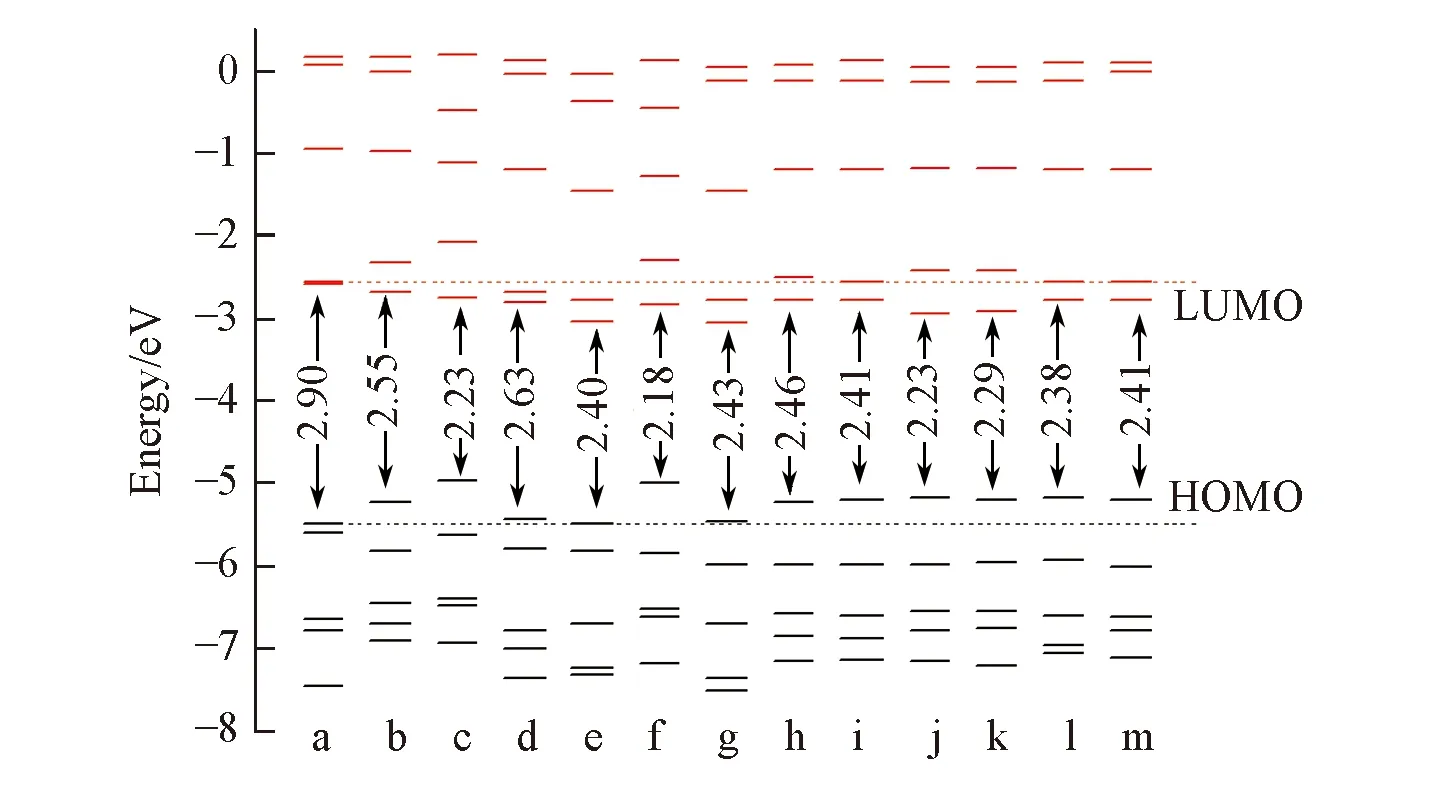
Fig.2 Orbital energy levels and the Egap of porphyrin and its isomersa. FBP; b. NECP; c. 2-NCP-2H; d. 2-NCP; e. 2,18-DNCP; f. 2,18-DNCP-2H; g. 2,12-DNCP; h. 1,7-Neo-C-NCP; i. 1,8-Neo-C-NCP; j. 1,12-Neo-C-NCP; k. 1,13-Neo-C-NCP; l. 1,17-Neo-C-NCP; m. 1,18-Neo-C-NCP.
前线分子轨道是阐明电子性质和解释电子跃迁的重要因素之一. 传统卟啉的电子光谱依赖于2个最高占据轨道(HOMO, HOMO-1)和2个最低未占据轨道(LUMO, LUMO+1)间的π→π*跃迁[36]. 对吸收光谱特征的理解和解释很大程度上与电子结构相关, 电子结构能够提供详细的轨道能量和组成. 根据分子轨道能级图(图2)可知, FBP具有最大的HOMO-LUMO的能级差(Egap), 氮杂卟啉的LUMO轨道能级均降低, HOMO轨道能级升高,Egap下降. FBP分子对称性高, 其LUMO和LUMO+1能级简并; 其它氮杂卟啉中因N/C交换导致分子对称性被破坏, 轨道简并消失. 单氮杂卟啉N/Neo-CPs分子的Egap都有所减小, LUMO/LUMO+1, HOMO/HOMO-1的能级差相应增大, 其中2-NCP-2H分子的Egap最小, LUMO/LUMO+1, HOMO/HOMO-1的能级差最大, 其Egap和FBP相比降低了0.67 eV. 双氮杂卟啉的Egap值进一步减小, 其中, 2,18-DNCP-2H的Egap值最小, 比FBP的小0.82 eV, LUMO/LUMO+1和HOMO/HOMO-1的能级差均增大, 电子跃迁方式增多; 双混氮杂卟啉Neo-C-NCPs中, 1,12-Neo-C-NCP的Egap最小, 比FBP的小0.67 eV. 虽然化合物2-NCP-2H和1,12-Neo-C-NCP的Egap值相同, 但其LUMO/LUMO+1, HOMO/HOMO-1能级水平不同, 电子跃迁吸收有差异, 表现为二者吸收光谱有差异.
占据分子轨道和未占据分子轨道的形状可协助直观分析卟啉分子的电子结构. 在FBP分子中, HOMO-1轨道主要分布在吡咯环的Cα和Cβ原子上, HOMO轨道主要分布在Cm和N原子上, LUMO轨道则分布在Cα和Cβ及Ⅰ和Ⅲ环的N原子上, LUMO+1轨道则分布在Cα和Cβ及Ⅱ和Ⅳ环的N原子上(图S4, 见本文支持信息). 氮杂卟啉由于原子位置的改变导致其分子轨道发生改变, 电子结构最大的变化在Ⅰ环(N/C交换的环), 在2-NCP-2H分子中, HOMO-1轨道在中心Ⅳ环C原子和Ⅲ环N原子上的分布增加; HOMO轨道在中心C原子(N/C交换的吡咯环)分布有所增加, 而在大卟啉环外的N和Cβ原子分布减小; LUMO轨道在卟啉中心的C原子分布有所增加; LUMO+1轨道在中心的C原子分布增加, 在Ⅱ/Ⅳ环的Cβ原子分布减小. 在双氮杂卟啉和双混氮杂卟啉中前线轨道分布也有类似的变化.
2.4 电子吸收光谱
已有实验测得苯溶剂条件下, FBP在396和564 nm处有吸收峰[35], 与本文计算所得苯溶剂中FBP在382和547 nm处的吸收峰基本吻合; 吸收光谱计算结果与Petit等[37]的计算结果相近, 可见, 本文采用的理论光谱计算方法是合理的.
真空条件下FBP的Soret带强吸收峰位于359 nm[摩尔吸光系数(ε)=250372.1]和375 nm(ε=161073.5), Q带无明显吸收峰. 与FBP相比, 单氮杂卟啉在Soret带吸收峰位置略微红移, 但在Q带出现吸收峰, 其中2-NCP-2H分子在Q带具有吸收振子强度最强且波长最长的吸收峰, 分别位于536和658 nm处, 其Soret带吸收峰位于375和395 nm处(图3). 双氮杂卟啉DNCPs化合物在Soret带的吸收峰处振子强度减小, Q带出现较明显吸收峰, DNCP-21H分子在Q带吸收峰分别位于566和669 nm处, Soret带吸收峰分别位于392和418 nm处, 与FBP谱峰相比分别红移了33和43 nm. DNCP类卟啉Soret带和Q带吸收峰均明显红移, 其中2,12-DNCP的Soret带吸收峰红移至466 nm处. 在双混氮杂卟啉Neo-C-NCPs中, 除1,7-Neo-C-NCP和1,18-Neo-C-NCP分别在564和568 nm处有宽吸收峰(含肩峰)外, 其它双混氮杂卟啉在Q带范围内均有2个吸收峰, 其中1,17-Neo-C-NCP在Q带最大吸收峰位于647 nm处. 光谱数据表明, N原子在不同位置对卟啉的吸收峰有不同影响. N原子位置的改变可导致卟啉化合物Soret带吸收谱峰增多且红移, Q带谱峰振子强度明显增强, 改善了卟啉在可见光区的吸收情况, 双吡咯N原子改变更加明显.

Fig.3 Simulated absorption spectra of porphyrins in vacuum

Fig.4 Hole-electron distributions of S1→S6 states in FBP, 2-NCP-2H, 2,18-DNCP-2H and 1,17-Neo-C-NCP The colors of blue and green indicate hole and electron distribution, respectively.
为更进一步理解电子跃迁情况, 利用Multiwfn分析分子的激发态, 通过空穴-电子分布图(等值线isovalue=0.002 eV)可直观地理解激发态电子跃迁, 图4为电子(蓝色)跃迁到空穴(绿色)部分示意图. FBP在Soret带359 nm处的吸收峰对应S0→S5跃迁, 而2-NCP-2H分子在Soret带的最大吸收峰375 nm对应S0→S6跃迁(图4和表S1, 见本文支持信息). 从空穴-电子分布图中电子和空穴的电荷质心的距离(DCT)数据可知, 在FBP中, 1~6个激发态的空穴和电子几乎是趋于卟啉大环的整体分布的, 电子跃迁没有明显的电子分布转移变化; 而在氮杂卟啉中, 1~6个激发态的空穴和电子分布有着分离的趋势, DCT值增加. 氮杂卟啉2-NCP-2H, 2,18-DNCP-2H 和 1,17-Neo-C-NCP的Q带吸收峰分别对应S0→S1和S0→S2跃迁, 其轨道跃迁组成相似, 但轨道跃迁贡献的差异导致其吸收光谱出现明显差异. 2,18-DNCP-2H在Q带669 nm处的吸收峰对应S0→S1跃迁以及对应的轨道跃迁贡献组成 HOMO-1→LUMO+1(4%)/HOMO→LUMO(94%); 1,17-Neo-C-NCP在Q带646 nm处的吸收峰也对应S0→S1跃迁, 但其对应的轨道跃迁贡献组成略有不同, 为HOMO-1→LUMO+1(8%)/HOMO→LUMO(88%)/HOMO→LUMO+1(3%).
2.5 溶剂效应
选择极性差异较大的水、 苯和氯仿3种溶剂, 研究了溶剂效应对卟啉能级轨道和吸收光谱的影响. 在13种卟啉中, FBP和1,12-Neo-C-NCP分子受溶剂影响最小, 在极性不同的溶剂中它们的Egap基本不变, 可能由于1,12-Neo-C-NCP分子的2个旋转吡咯环处于对位, 分子电荷总体上仍呈一定的对称性, 受溶剂极性影响较小所致. 2-NCP, 2,18-DNCP-2H和1,13-Neo-C-NCP分子随着溶剂极性减小Egap均略微增大(图5、 图S5和图S6, 见本文支持信息). 除上述分子外, 卟啉类化合物的Egap随着溶剂极性减小而减小, 化合物吸收峰红移. 随着溶剂极性减小, FBP的LUMO+4至LUMO+6轨道能级升高, 而LUMO至LUMO+3和HOMO至HOMO-6轨道能级降低, 轨道能级的改变导致其吸收光谱的变化. 在极性较小的苯溶剂中FBP的 Soret带吸收峰处振子强度值最大, 峰位比真空条件Soret带吸收峰红移23 nm, 在溶剂条件下其Q带在540~550 nm附近出现了微弱的吸收峰.
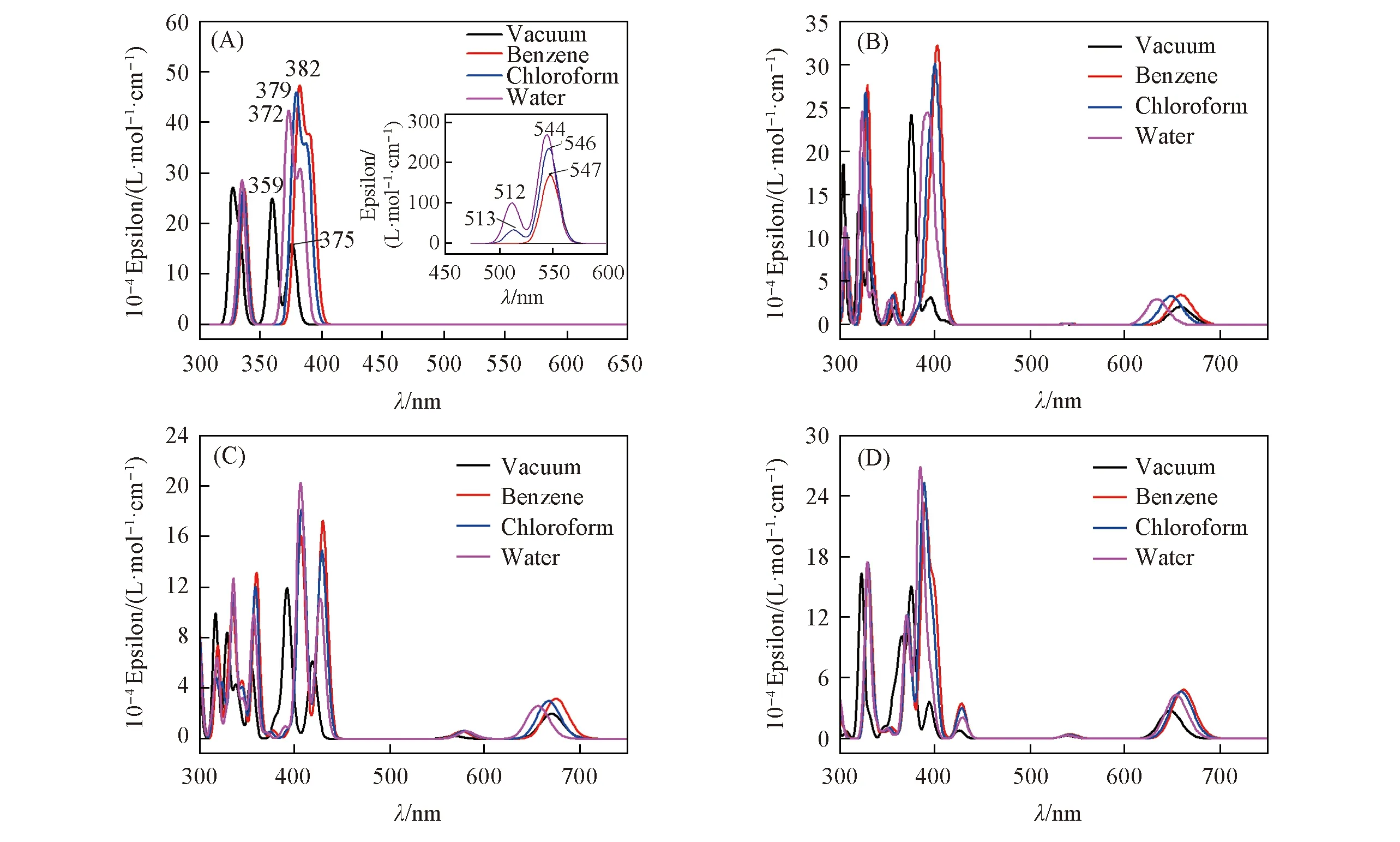
Fig.5 Simulated absorption spectra of FBP(A), 2-NCP-2H(B) , 2,18-DNCP-2H(C) and 1,17-Neo-C-NCP(D) in vacuum, benzene, chloroform and water
随着溶剂极性减小, 单氮杂卟啉中2-NCP-2H的LUMO+3至LUMO+6轨道能级升高, LUMO至HOMO-6轨道能级降低,Egap随着溶剂极性的减小而减小, Soret吸收峰随着溶剂极性的减小而红移; Q带吸收峰受溶剂影响更为敏感, 苯溶剂中最大吸收峰红移25 nm. 1,17-Neo-C-NCP分子Soret带吸收峰随着溶剂极性减小吸收峰红移, 但在弱吸收峰425 nm位置未发生明显变化; Q带吸收峰随着溶剂极性减小而红移, 振子强度增加, 在不同溶剂中的最大位移差为8 nm. Q带吸收峰主要是由S0→S1和S0→S2跃迁引起的, FBP分子的S0→S1和S0→S2跃迁分别由HOMO-1→LUMO(41%)/HOMO→LUMO+1(58%)和HOMO-1→LUMO+1(46%)/HOMO→LUMO(54%)组成, 氮杂卟啉2-NCP-2H, 2,18-DNCP-2H和1,17-Neo-C-NCP的S0→S1跃迁的主要轨道组成分别为HOMO→LUMO(94%)和HOMO→LUMO(88%), S0→S2跃迁的主要轨道组成分别为HOMO-1→LUMO(44%)/HOMO→LUMO+1(55%), HOMO-1→ LUMO(44%)/HOMO→LUMO+1(55%)和HOMO-1→LUMO(36%)/HOMO→LUMO+1(60%). Soret带吸收峰主要是由S0→S5和S0→S6跃迁引起的, FBP分子的S0→S5跃迁是由HOMO-3→LUMO(21%)/HOMO-1→LUMO+1(40%)/HOMO →LUMO(36%)组成, 2-NCP-2H的S0→S5跃迁主要由HOMO-3→LUMO(85%)组成.
3 结 论
单、 双及双混氮杂卟啉在Q带光吸收性质差别较大, 原因在于N原子位置改变导致卟啉分子轨道能级的变化, 卟啉大环结构平面具有不同程度扭曲, 共轭程度改变,Egap减小, 部分电子禁阻跃迁转变为弱允许跃迁, 氮杂卟啉的电子跃迁途径增多, 跃迁几率增加, 进而对分子吸收光谱产生影响. 与对称性较高的FBP相比, 氮杂卟啉化合物在N原子位置改变后, HOMO轨道能级升高、 LUMO轨道能级下降, 即能级差减小, 吸收光谱红移. 单氮杂卟啉2-NCP-2H、 双氮杂卟啉2,18-DNCP-2H和双混氮杂卟啉1,17-Neo-C-NCP的能级差降低最多, 谱峰红移更为显著, 可作为新型可见光吸收材料的母核结构进一步修饰开发. 溶剂对氮杂卟啉光谱影响明显, 随着溶剂极性减小, N-/Neo-CPs, DNCPs与Neo-C-NCPs的Soret带和Q带吸收峰的位置发生红移越显著. 可为筛选以卟啉环或氮杂卟啉环作为母环合成新型敏化剂材料提供重要理论依据.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20190413.