Understanding and Applying the Length-scale Dependence Mechanism of Hydrophobic Interaction
2020-02-12DiWeiShuaiWangJuanMeiYueHaiCaoYi
Di Wei-Shuai ,Wang Juan ,Mei Yue-Hai ,Cao Yi ,2,3∗
1Collaborative Innovation Center of Advanced Microstructures,National Laboratory of Solid State Microstructure,Department of Physics,Nanjing University,Nanjing 210093,China
2Institute for Brain Sciences,Nanjing University,Nanjing 210023,China
3Key Laboratory of intelligent Optical Sensing and Integration,Nanjing University,Nanjing 210023,China
Abstract:The length-scale dependence theory established by Lum,Chandler and Week provided a new insight into interpreting,studying and applying hydrophobic interaction.Hydrophobic hydration free energy decreases with the volume of solute at small length regime(<1 nm)and increases with the surface area of solute at a larger length regime(>1 nm).The crossover length near 1 nm indicates an entropic and enthalpic co-dominant hydration process at micro level which is distinct to the macro-level phenomenon.In this review,we showed the dependence of hydrophobic hydration free energy on temperature,pressure and additives.Especially,single-molecule force spectroscopy was introduced to provide a method to investigate hydrophobic interaction with respect to polymers.Furthermore,various applications relevant to hydrophobic interaction were represented to inspire deeper researches and explorations.Moreover,hydrophobic interaction underlying the protein folding and membrane assembly was further understood based on these promising progresses.
Key Words:Hydrophobic Interaction;Length-scale Dependence;Single-Molecule Force Spectroscopy;Driving Force;Protein Folding;Membrane Assembly
CONTENTS
IV.Perspective and Future 15
I.Introduction 1
II.Theory 2
A.LWC Length Scale Dependence Theory 2
B.Temperature Dependence of Hydrophobic Hydration Free nergy 3
C.Pressure and Additives Dependence of Hydrophobic Hydration Free Energy 4
D.Experimental Development for the LWC Theory 5
III.Application of Hydrophobic Interactions. 8
A.Hydrophobic Driving Force 8
B.Entropy Convergence in Protein Folding 12
C.Protein Folding and Membrane Self-assembly 12
References 15
I.INTRODUCTION
Hydrophobic interaction has now drawn great research interest due to its significant roles in many biological processes,including protein folding,membrane formation and phase separation.At macroscopic level,hydrophobic interaction leads to phase separations between oily substances(such as toluene,benzene,dichloromethane)and water.The bulk oily substances scattering in the water will shape into small globules and form emulsion to minimize the interfacial energy and maintain the stability of the system.
At microscopic level,it is widely known that water is a polar molecule.Most polar molecules(for instance,ethyl alcohol,formaldehyde and acetic acid)are soluble in water.In contrast,nonpolar molecules(for example,toluene,alkane and ethyl acetate)tend to aggregate together in water.The force that drives such phenomenon is the so-called hydrophobic interaction.
Physically,hydrophobic interaction is a long range interaction in a wide distance up to 20 nm,much longer than a single covalent bond,and decays exponentially[1].Generally,hydrophobic interaction is comparable to common weak molecular interactions,such as hydrogen bond,van der Waals force,cation-π interactions and halogen bond,but much weaker than the covalent bond,ionic bond and metallic bond.
It is universally accepted that hydrophobic interaction plays a significant role in the folding and assembly of various proteins,the formation of biological membrane as well as micelle and ligand-receptor binding[2−9].By investigating hydrophobic interaction,a deeper understanding of the life processes at the molecular level can be obtained.
In this review,we discuss the current understanding and the evolution of the theory on hydrophobic interaction.The experiments and simulations that support these discoveries are summarized.Single-molecule force spectroscopy is also introduced as one of the most efficient tools to study hydrophobic interactions at the molecular level.Moreover,various applications of the theories are outlined to pave a way for future research.
II.THEORY
A.LWC Length Scale Dependence Theory
There were a lot of efforts on the theory of hydrophobic interaction.Tolman length[10],the scaled particle theory and its revised version[11−13],and the information theory[14]influenced the research of hydrophobic hydration for half a century.However,these methods and theories failed to explain some other experimental phenomenon more or less.For instance,temperature dependence of hydrophobic hydration free energy was missed in the scaled particle theory.
At present,length scale dependence theory(the LCW theory),first established by Lum,Chandler,and Weeks,is the most widely accepted one in the field of hydrophobic interaction research[15].The LCW theory is beginning at the view of local density fluctuation to estimate the hydrophobic hydration free energy of hydrophobic species with treating solvent-solute attraction as first order perturbations[16−20]or via mean field theory[21].Considering the hydrophobic species as a hard sphere,the hydrophobic hydration free energy(∆G)for a hydrophobic solute with the excluded volume(v)can be written as:
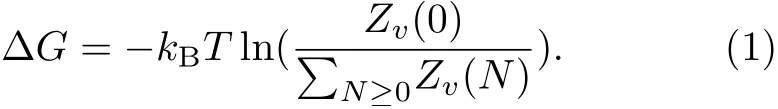
Here,kBis the Boltzmann constant,T is temperature in Kelvin and Zv(N)is the partition function when N solutes occupy the volume(v)that can be estimated by the Gaussian statistics.

FIG.1.The hydrophobic hydration free energy per surface area(∆G/4πR 2)exhibits a function of radius of hydrophobic species based on the hard sphere model at ambient conditions.Two black lines correspond to the fitting of the crossover behavior.γ is the surface tension of bulk water.The data was adapted with permission from David M.Huang,Phillip L.Geissler and David Chandler,J.Phys.Chem.B,2001,105:6704.Copyright(2001)American Chemical Society.
The final estimating result of∆G indicates that the hydrophobic hydration free energy per surface area is proportional to the radius of sphere when the radius is smaller than one nanometer and converges to a constant value at a larger radius of several nanometers or more.This converged value can be recognized as the surface tension of bulk water[Fig.1].It is worth to pointing out that this crossover behavior does not correspond to a phase transition but involves two different hydrophobic hydration mechanisms.
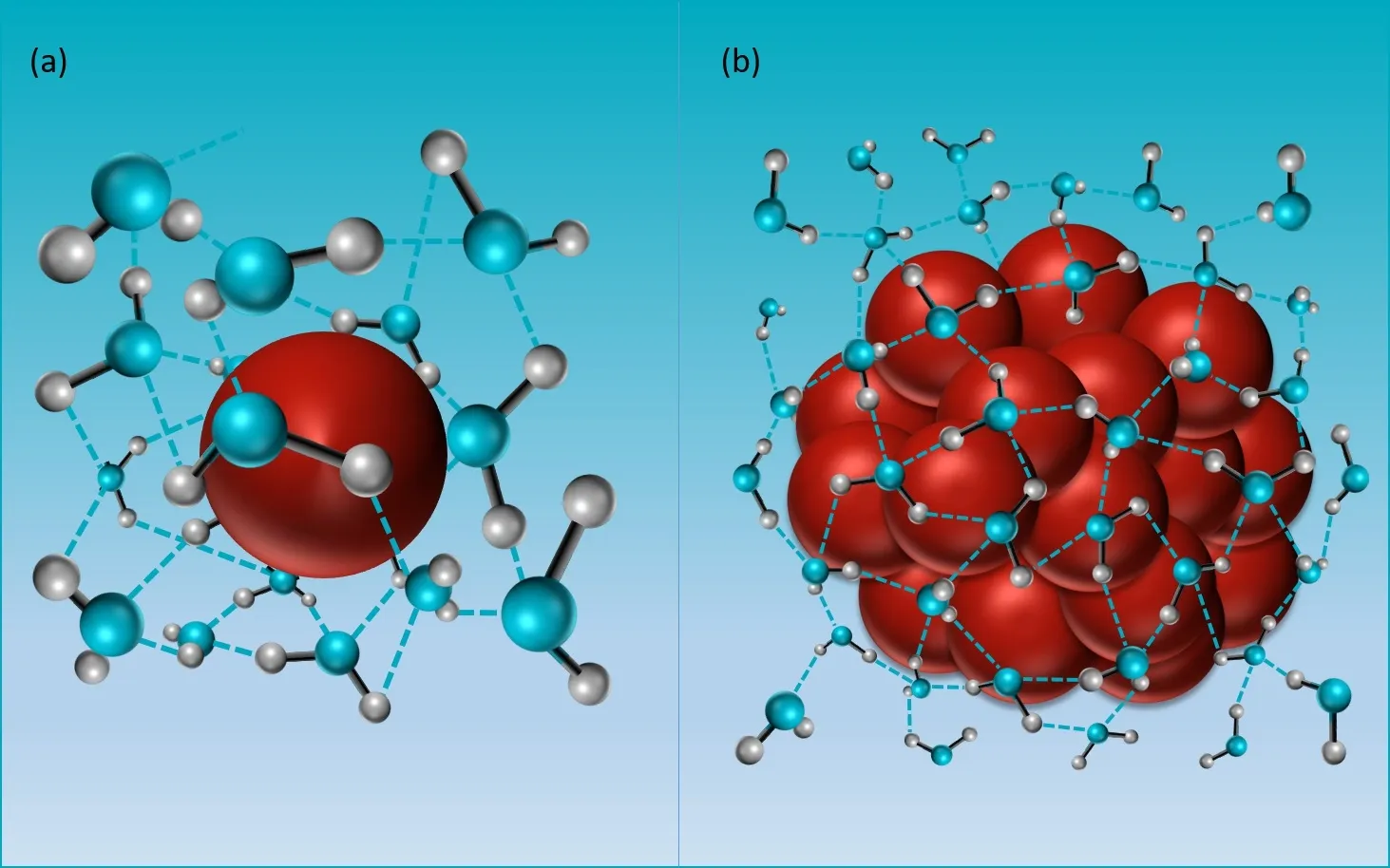
FIG.2.Molecular dynamic simulations of the hydrophobic hydration of small solutes and large solutes.The red sphere represents the hydrophobic solute and the water molecule is depicted by the one oxygen atom(blue)bonding with two hydrogen atoms(gray).(a)A small hydrophobic solute can be held in a cavity-like region formed by the spatially adjacent hydrogen bonds.(b)Many previous hydrophobic solutes aggregate into a much larger hydrophobic solute that cannot be accommodated by the cavity formed by the hydrogen bonds.As a result,hydrogen bonds are distorted and water molecules rearrange around the surface of newly formed larger solute,resulting in a dewetting interface.The picture was adapted from Chandler[3].
As we all know,the bulk water contains countless hydrogen bonds,forming a complex but orderly hydrogen bond network.Spatially adjacent hydrogen bonds can form cavity-like region with an excluded volume in the network[3,22].
The currently widely accepted idea that the small hydrophobic solutes would be accommodated in the cavities formed by the hydrogen bonds[Fig.2(a)]and the larger hydrophobic solutes would distort the hydrogen bond network around the solute surface,was first proposed by Stillinger in 1973[12]and later improved by Chandler after 30 years[3,22][Fig.2(b)].Inclusion of small hydrophobic solutes gives rise to a spatial rearrangement of hydrogen bonds surrounded around the hydrophobic particles.The hydrophobic hydration free energy is dominated by the entropic change of the whole system due to inserting hydrophobic particles in the cavity-like region.In contrast,the existing hydrogen bonds are not able to maintain previous state around the larger hydrophobic solutes surface,resulting in exclusion of local water molecules moving away from the surface of the hydrophobic solutes.Such process that involves breaking the hydrogen bonds and forming an interface between the water molecules and hydrophobic solutes will bring an obvious enthalpic change to the system as well as an entropic change.However,the enthalpic change is dominant in this process.Moreover,this newly formed interface represents the dewetting of hydrophobic solutes[3,23].
B.Temperature Dependence of Hydrophobic Hydration Free nergy
At ambient conditions,crossover length of hydrophobic hydration free energy is about 1 nm,and the converged value is near surface tension of bulk water.Experiment confirms that the surface tension of water decreases with increasing temperatures.Consequently,it is easy to propose that the change of external conditions is able to change the dependence behavior of the hydrophobic hydration free energy.
Huang and Chandler first applied the LCW theory to calculate the hydrophobic hydration free energy of the hard sphere at different temperature[Fig.3][22].When the radius of sphere is smaller than 10 angstroms,the hydrophobic hydration free energy per surface area is almost not affected by the temperature in the range from 277 K to 423 K[Fig.3(a)].Furthermore,the crossover length is independent on the temperature in this range.In contrast,the hydrophobic hydration free energy per surface area monotonically decreases with increasing temperatures when the radius is larger than 10 angstroms.Calculations indicate that the descending converged value is in accordance with the decreased surface tension of bulk water caused by increased temperature.Therefore,the temperature influence of hydrophobic hydration free energy mainly works on the enthalpic-dominated process at large length regime.
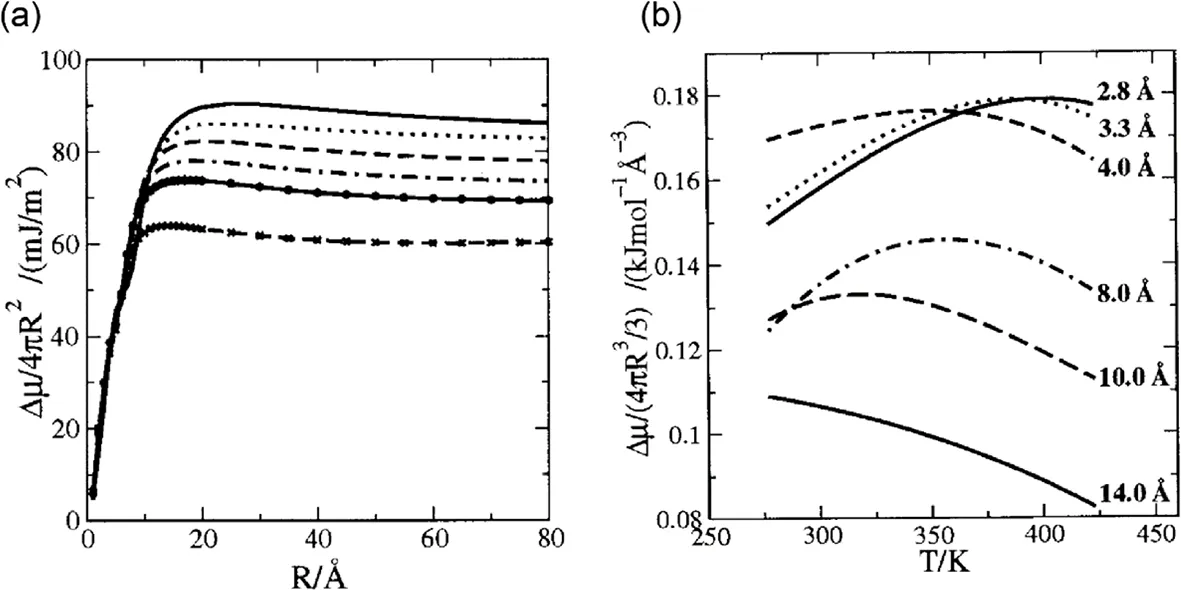
FIG.3.The influence of temperature on the hydrophobic hydration free energy.(a)The relationship between hydrophobic hydration free energy per surface area and the radius of the hard sphere at different temperatures.From the top down,the temperature is 277 K,298 K,323 K,348 K,373 K and 423 K,respectively.(b)The relationship between hydrophobic hydration free energy per volume and temperature at different radius.The data was adapted from Huang and Chandler[22].Copyright(2002)National Academy of Sciences.
Fig.3(b)shows a no-monotonous relationship between the hydrophobic hydration free energy per volume and the temperature.When the radius is smaller than 10 angstroms,the hydrophobic hydration free energy per volume reaches a maximum.Furthermore,this maximum shifts to left with increasing radius smaller than 10 angstroms.Relevant experiments of small molecules,including methane,neon,argon and xenon had verified this simulated results[19]. Besides,the maximum vanishes and the curve exhibits a monotonic decrease.This phenomenon can be interpreted that the thermodynamic process of cavity holding small solute is dominated by the entropy with a certain contribution of enthalpy.However,the enthalpy is greatly affected by the temperature than entropy.With the increasing temperatures,enthalpy plays a more and more powerful influence on the hydrophobic hydration free energy,thus leading a maximum.The larger solute needs a slightly higher temperature,as a result of which,the maximum shows a left shift when the solute gets bigger within 10 angstroms.
It is worth mentioning that for small sphere(radius less than 1 nm),the entropic change of system undergoes from a negative value to a positive value.That means,at a certain temperature,the entropic change of system is zero,similar to the concept of entropy convergence in protein folding[24].
Because of∆S=−(∂(∆G)/∂T)P(the subscript P denotes as the constant pressure),the temperature corresponding to the maximum in Fig.3(b)just indicates the position of zero of the entropic change.When higher than this temperature,the entropic change is always similar to the condition of larger spheres.When lower than this temperature,taking account for∆G=∆H−T∆S,the term−T∆S can maintain an approximately unchangeable value owing to the decrease of term−∆S with simultaneously increasing term T.It is the reason of that temperature does not obviously affect hydrophobic hydration free energy at small length regime in Fig.3(a).
C.Pressure and Additives Dependence of Hydrophobic Hydration Free Energy
Computation and thermodynamic analysis revealed the different mechanism of temperature dependenceof hydrophobic hydration freee nergy.How about pressure?
Garde and coworkers investigated the influence of the hydrophobic hydration free energy brought by external pressure[25].By applying negative pressure,the system was under tension rather under stress.With increasing tension,the hydrophobic hydration free energy decreased[Fig.4(a)].
According to authors’view,the LCW theory is based on the local density fluctuations to calculate the hydrophobic hydration free energy[15].Under lager tension,microscopic water molecular structure would change.More specifically,dilution of water molecules adjacent to solutes leads to the decrease of local density.Therefore,hydrophobic hydration free energy decreases with the increasing negative pressure.
Besides,the crossover length would shift to a smaller value with an increasing negative pressure,even to 0.3 nm at−1,000 atm,which is a distinct picture compared to the temperature dependence in Fig.3(a).The critical point of understanding this picture lies in the equation∆G=∆H−T∆S,with the help of the LCW theory. At small length regime,term−T∆S dominates the term∆G. As we all know,(∂S/∂P)T=−(∂V/∂T)P(the subscript T and P denote as the constant temperature and pressure,respectively)from thermodynamic Maxwell relation.Because of the small isobaric expansion coefficient of water,the entropic change of system is not sensitive to the pressure change at a constant temperature.In contrast,enthalpic change,∆H is directly affected by the pressure change as seen by equation H=U+PV.Collectively,with increasing negative pressure,term−T∆S gradually loses the dominant position in the term∆G compared to previously minor term∆H.As term∆H plays a more and more important role in term∆G,one can image that the crossover length will become more and more smaller even to vanish at a much higher temperature.
Subsequent simulated computation by Garde and coworkers had verified above analysis.They carried out simulations to separately compute the term∆H and term−T∆S in term∆G at 1 atm and−1,000 atm at temperature 300 K[Fig.4(b),(c)].At 1 atm,Fig.4(b)clearly shows that term−T∆S takes over predominant state in term∆G as predicted by the LCW theory.Nonetheless,when at−1,000 atm,term−T∆S becomes less important especially beyond 0.3 nm and term∆H undertakes the major change in term∆G.
Next,they studied the influence of the hydrophobic hydration free energy brought by additives[Fig.4(d)][25].Addition of sodium chloride increases the hydrophobic hydration free energy and the higher concentration makes the more obvious effects.In contrast,addition of ethanol shows an opposite trend.At larger length regime,increased hydrophobic hydration free energy per surface area is consistent with the increased water surface tension due to the addition of salt.Similarly,the addition of ethanol also follows this mechanism but in an opposite direction.
Inset in Fig.4(d)shows a similar result that entropic change term no longer occupies the dominant position in the whole free energy change with addition of 40 mol%ethanol compared to Fig.4(c).The sole difference is that enthalpic change dominates the change of hydrophobic hydration free energy from the beginning in Fig.4(d)inset.Correspondingly,the result with additives separates from water’s result beginning at small length regime.A speculation at the molecular level is that additives would change the previous hydrogen bond network,resulting in no cavity or much smaller cavity to accommodate the hydrophobic solute.Consequently,crossover length gradually becomes smaller(addition of ethanol)or vanishes(addition of sodium chloride).
D.Experimental Development for the LWC Theory
More and more theoretical and simulated researches threw a profound light on hydrophobic hydration for us,however,experimentally direct calculation of hydrophobic hydration free energy is lack for all time.
In fact,many experimental efforts focusing on calculating hydrophobic hydration free energy were done by lots of researchers but fruitless.Because of insolubility of hydrophobic molecule,it is experimentally difficult to get values from solubility data.Early method to quantify hydrophobic hydration free energy was to measure transfer free energy from water to a nonpolar reference solvent(such as a liquid alkane)[11,27−29].However,values obtained by this method were controversial even not correct[30−32]and had been in conflict with subsequent successful small-to-large length scale theory[15].Afterwards,Ben-Amotz exerted Raman scattering method to directly measure hydrophobic hydration shell[33]but this kind of method cannot get insight into hydrophobic molecule.

FIG.4.Influence of pressure and addition of salt and ethanol on hydrophobic hydration free energy.(a)Hydrophobic hydration free energy per surface area v.s.radius at different pressure.Data of black dots was adapt from Chandler[26].(b)and(c)Computational simulations on entropic change and enthalpic change at 1 atm and−1,000 atm,respectively.(d)Hydrophobic hydration free energy per surface area v.s.radius with different additives.Inset:Computational simulations on entropic change and enthalpic change with 40 mol% ethanol.The data was adapted from Garde[25].Copyright(2005)National Academy of Sciences.
Recently,Li and Walker initiated a novel design to investigate hydrophobic polymers by single-molecule force spectroscopy based on atomic force microscopy(AFM)[34−37].AFM is a powerful tool to study diverse issues in biophysical and chemical fields at single cell or molecular level[38−46].In a typical single-molecule force spectroscopy experiment,a target molecule chemically or physically linked between the substrate and AFM cantilever tip is stretched until the specific chemical bond or absorption ruptures. Resultant forceextension curves provide an insight into energetic information of single molecule that includes dissociation forces and rates,intermediated states,changes of conformations,free energy and so on.
Polystyrene with a molecular weight about 100 kDa was used in Li and Walker’s experiment.In a chain of polystyrene,each monomer is a hydrophobic unit.Due to hydrophobic hydration[3,47−49],all the hydrophobic monomers will aggregate together and hence polystyrene chain will collapse into a compact coil in water which can be modeled as a hard sphere[34].Fig.5 shows an AFM picture of polystyrene that collapses into a sphere verifying the above analysis of hydrophobic collapse.The polystyrene was attached to the silicon wafer via chemical linkage and then immersed into water to make it collapse into sphere[50].
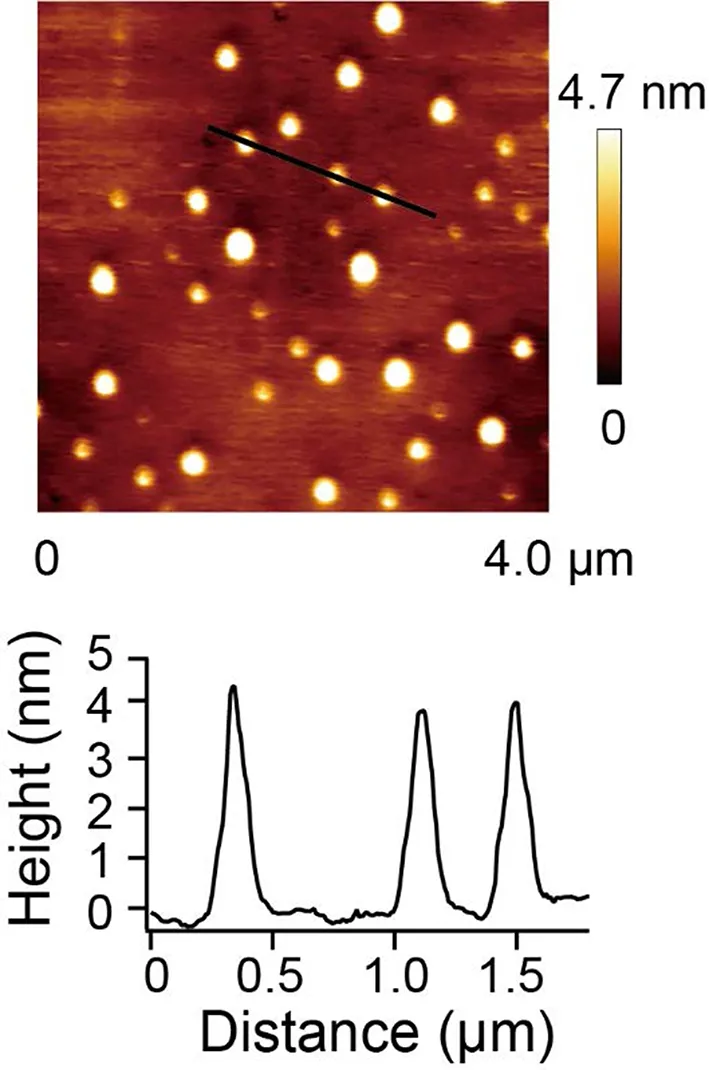
FIG.5.AFM image of a single polystyrene chain collapsed into a sphere.The lower panel corresponds to the particles signed by the black line.The picture was adapted from Cao et al.[50],Reprinted figure with permission from Yi Cao et al.,Phys.Rev.Lett.,2019,122:047801.Copyright(2019)by the American Physical Society.
Li and Walker employed the single-molecule force spectroscopy experiments to unfold a polystyrene chain which had collapsed into a compact sphere in water[Fig.6(a)]. From beginning to end,polystyrene nanosphere undergoes fully collapsed state,collapsedextended coexisted state and fully extended state represented by upper panel in Fig.6(b).Force-extension curve shows a typical force plateau with a subsequent elastic stretching[Fig.6(b)].The force plateau corresponds to the process that a polystyrene nanosphere gradually changes into a polystyrene chain under stretching.The elastic stretching refers to the process that a polystyrene chain is pulled which behaves like a polymer in good solvent that can be modeled by Worm-like chain model[Fig.6(b)][51]. During the whole unfolding process,the radius of polystyrene nanosphere changes from initial several nanometers to later few nanometers,indicating that the process of unfolding polystyrene nanosphere involves two different hydrophobic hydration mechanisms.It is such a novel design that offers a unified view in investigating hydrophobic hydration free energy at experimental aspects.
Fig.6(b)and 6(c)provide an intuitive assay to directly calculate hydrophobic hydration free energy from experimental data.External work done by stretching force that transforms a polystyrene from a collapsed state to a fully extended state is∫Fdz.F is the plateau force and integral region is extension that corresponds to force plateau.In other words,∫Fdz equals to a rectangle area encircled by plateau force and extension.Stretching process causes a decrease in conformational entropy denoted as T∆Sext(∆Sext<0)that can be calculated by the integration of blue region under Wormlike chain fitting in Fig.6(b).Besides,supposing that hydrophobic hydration free energy of each monomer is identical denoted as∆G,thus the hydrophobic hydration free energy of a whole chain is N∆G(N is the number of monomers).In fact,value of N∆G corresponds to integration of red region in Fig.6(b).To sum up,equation
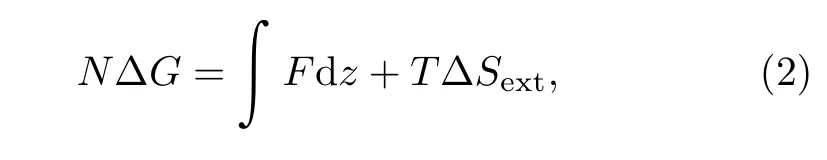
concludes a free energy change in a single-molecule experiment.
Although Li and Walker’s work acquired a pretty good achievement,especially for that they obtained a temperature dependence of hydrophobic hydration free energy on various polymers(besides polystyrene,including poly-4-tert-butylstyrene and poly-4-vinylbiphenyl)which provided an insight into influence of side groups on hydrophobic hydration free energy[35],they failed to get a direct length scale dependence of hydrophobic hydration free energy predicted by the LCW theory based on their theoretical framework.
As mentioned above,hydrophobic hydration free energy of each monomer of polymer(∆G)was identical and the whole energy was N∆G in Li and Walker’s theoretical framework.That means interactions among monomers were ignored which equaled to neglecting polymerization effect in lower panel in Fig.6(c).More importantly,conformational entropy was considered while a characteristic elastic entropy of extended polystyrene chain that behaved like a polymer in good solvent was lost.
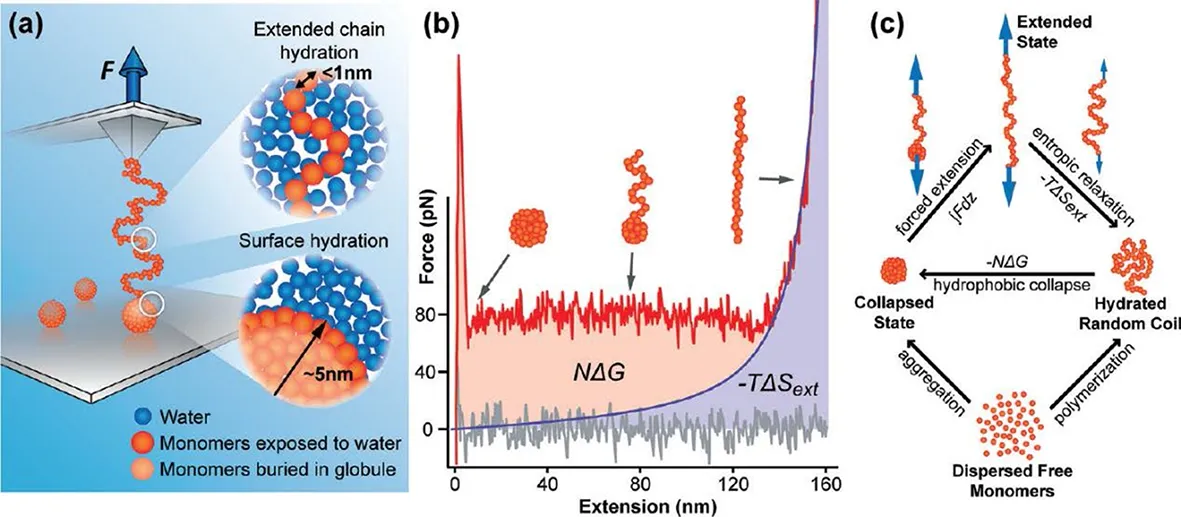
FIG.6.(a)Schematic of single-molecule force spectroscopy experiment on unfolding a polystyrene nanosphere.(b)Typical force-extension curve of unfolding a polystyrene nanosphere.The nanosphere undergoes initial collapsed state,intermediate collapsed-extended state and final fully extended state(upper panel denoted by black arrow).Blue curve is fitted to elastic stretching part by Worm-like chain model.(c)Schematic of free energy change among various conformational state.The picture and data were adapted with permission from Isaac T.S.Li and Gilbert C.Walker,Accounts.Chem.Res.,2012,45:2011.Copyright(2012)American Chemical Society.
Hence,Cao and coworkers further developed Li and Walker’s work and put forward a new theoretical approach to calculating the hydrophobic hydration free energy of polystyrene nanosphere[50].
During stretching process,given that the total free energy of system,G(x,L,R),can be written as

Here,R is the radius of collapsed sphere,x is extension and L is the contour length of extended chain.Gsph(R)is target expression that represents the free energy of collapsed sphere.Gchain(L)represents the free energy of extended chain and GWLC(x,L)represents the entropic energy of extended chain of Wormlike chain fitting(Fig.7(a)).
With the exception of conventional single-molecule experiments,they also carried out refolding experiment on stretching polystyrene nanosphere.Superimposed curves indicate that unfolding polystyrene nanosphere is a reversible process[Fig.7(b)].
Based on this,they used Lagrange multiplier method to find the extreme point of the total free energy,G(x,L,R)with two constraints to solve the target expression Gsph(R).The final expression of Gsph(R)is written as:
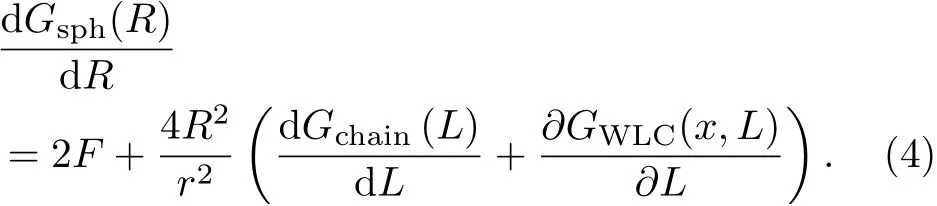
Through numerical integral calculation,it is easy to obtain the change of free energy of polystyrene nanosphere,∆Gsph(R).Fig.7(c)and 7(d)show that hydrophobic hydration free energy and its per surface area as a function of radius of nanosphere,R,respectively.Surprisingly,the latter shows a crossover behavior that is in agreement with the LWC theory’s prediction.To a certain extent,it is the first direct experimental verification for the LWC theory.
III.APPLICATION OF HYDROPHOBIC INTERACTIONS.
A.Hydrophobic Driving Force

FIG.7.(a)Schematic illustration of the theoretical model.During stretching process,collapsed part is regarded as a hard sphere with radius,R,and extended chain is regarded as a rod with contour length,L and radius,r.Note that r is the scale of a styrene molecule.X is the distance between AFM tip and the substrate.There are two constraints in this model used in Lagrange multiplier method:total volume of collapsed sphere and extended chain is constant,and distance between tip and substrate,X,equals to extension x plus diameter of sphere,2R.(b)single-molecule force spectroscopy experiment on refolding PS nanosphere.1:extend,2:half-retract,3:re-extend,4:full-retract.(c)Hydrophobic hydration free energy as a function of radius of polystyrene nanosphere.(d)Hydrophobic hydration free energy per surface area as a function of radius of polystyrene nanosphere.The picture and data were adapted from Cao et al.[50],Reprinted figure with permission from Yi Cao et al.,Phys.Rev.Lett.,2019,122:047801.Copyright(2019)by the American Physical Society.
As reviewed above,hydrophobic hydration free energy of small solute is proportional to the excluded volume of solute.For sufficiently large solute,hydrophobic hydration free energy is proportional to the surface area of solute.Imaging that there are N identical small hydrophobic particles and each hydrophobic hydration free energy is denoted as∆GV.As a result,the whole hydrophobic hydration free energy is N∆GV.On the other hand,if the N particles aggregate into together to form a large particle whose radius outnumbers the crossover length,then the hydrophobic hydration free energy is proportional to the newly formed surface area denoted as∆GS.Generally speaking,the N∆GVis larger than the∆GSand the GD=N∆GV−∆GScan be regarded as the driving force for assembly of the hydrophobic particles[3].
As showed in Fig.8,the larger N makes a larger driving force(GD)within a certain length range.In addition,driving force will become larger at a higher temperature.There are two reasons for that.On one hand,a higher temperature results in a lower surface tension(γ)indicated by a lower horizontal line at large length regime.On the other hand,entropic dominated process ispositively related to temperature(∆G=∆H−T∆S;at small length regime,∆S(=−(∂(∆G)/∂T)P)is negative[22])indicated by a sharper sloping line at small length regime.
Upon protein folding,hydrophobic driving force is a result of ensemble interactions between water and hydrophobic amino acid residues.As hydrophobic driving force is influenced by the temperature,Abeln and coworkers investigated the temperature(265 K∼340 K)dependence of hydrophobicity of various types of amino acid residues based on a large set of protein structures(form PDB)using nuclear magnetic resonance[52].
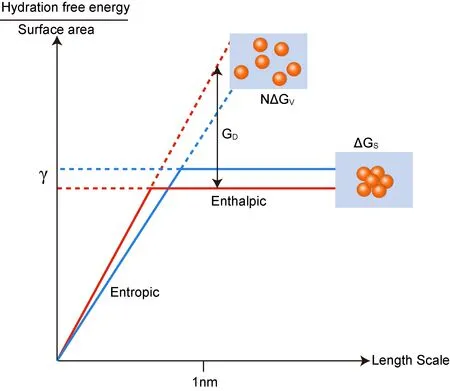
FIG.8.The schematic of driving force,G D,for small hydrophobic particles assemble into a large compact cluster.Sloping line and horizontal line represent the different hydrophobic hydration mechanism mentioned previously,respectively.In addition,the red lines indicate the result of higher temperature.Correspondingly,the blue lines indicate the result of lower temperature.The picture was adapted from Chandler[3].
Generally speaking,the hydrophobic amino acid residues are buried into the inner space of protein away from water. The authors classified 20 amino acids into five types,including the hydrophobic(Ala,Ile,Leu,Met,Val),the aromatic(His,Phe,Trp,Tyr),the charged(Arg,Asp,Glu,Lys),thepolar(Asn,Gln,Ser,Thr)and the other(Cys,Gly,Pro)type.The transfer free energy for hydrophobic amino acid residues from the inner space of protein to surface facing the water was calculated through three different methods that provided consistent results.Estimates indicated that the transfer free energy of the hydrophobic type and the aromatic type showed an obvious non-monotonic temperature dependence with a maximum at around 300 K which was similar to the relationship in Fig.3(b).In contrast,the transfer free energy of other three types showed little temperature dependence.
In order to further study the temperature dependence,Abeln and coworkers calculated transfer free energy of individual amino acid residue within hydrophobic and aromatic types.Except for His,the transfer free energy of the other eight amino acid residues showed also an obvious non-monotonic temperature dependence with a maximum at around 300 K that was similar to the above results of hydrophobic and aromatic types.
Overall,their findings showed that hydrophobic interactions in water-protein system became weaker at a lower temperature compared with a higher temperature which was in accordance with the previous theoretical predictions[3].Furthermore,this can be used to interpret that some proteins unfold at low temperature(cold denaturation)[53,54].
Afterwards,Vendruscolo and coworkers investigated the difference of protein structure between hot(323 K)denatured state and cold(272 K)denatured state compared with the nature(298 K)state via restrained molecular dynamics simulations and replica-averaged metadynamics[55].
Yeast frataxin was used in their simulations for the observation of its hot and cold denatured states at neutral pH without adding destabilizing agents.The authors found that the cold denatured state was more expanded than hot denatured state in structure which can be evidenced by the radius of gyration.The radius of gyration at the cold denatured state,hot denatured state and nature state was 1.7 nm,1.6 nm and 1.5 nm,respectively.Besides,the hot denatured state was ampler in content of α-helical and β-sheet than the cold denatured state.Overall,the hot denatured state was more“functional”than cold denatured state in structure.
The difference of three states in structure suggested the different properties of corresponding three water-protein systems.Therefore,the authors studied the number of hydrogen bonds of water-protein system.The number of hydrogen bonds per water molecule including water-water hydrogen bond and water-protein hydrogen bond is nothing different in the bulk water(BWM)with regard to the interface of the first hydration shell(IWM)[Fig.9(a),(b),(c)].However,the number of water-water hydrogen bonds per water molecule decreases with increasing temperature while keeping with a constant number of water-protein hydrogen bonds.This is consistent with previous research result[56−58].Further analysis indicates that protein prefers to form hydrogen bond with water molecule compared to other proteins[Fig.9(d)]at the denatured state.That is to say the interaction between protein and water becomes stronger which is a clue to expanded structure at lower temperature.By contrast,at nature state,protein inclines to form hydrogen bond with protein rather than water,resulting in a more compact structure[Fig.9(e)].However,formation of hydrogen bond does not show preference between water and protein at hot denatured state[Fig.9(f)].Besides,the most protein-protein hydrogen bonds formed at nature state compared with cold denatured state suggesting that hydrophobic driving force promotes the stability of protein.When it comes to hot denatured state,larger hydrophobic driving force is offset by thermal perturbations and thus leads to an intermediately stable structure.
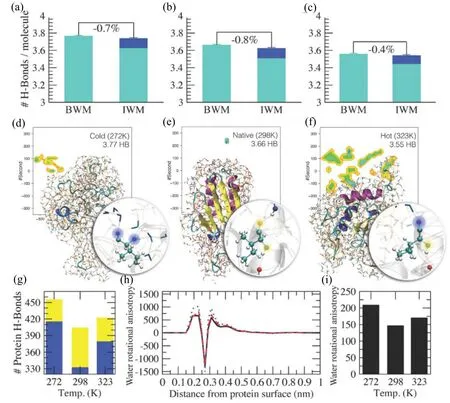
FIG.9.Characterization of hydrophobic effect upon hydrogen bond.(a),(b)and(c)The number of hydrogen bonds per water molecule in the bulk water(BWM)and the interface of first hydration shell around the protein(IWM)at different temperature(Donated as the cold denatured state,nature state and hot denatured state,respectively).The light blue and blue denote the water-water hydrogen bond and water-protein hydrogen bond,respectively.(d)At the cold denatured state,water molecule is prior to form hydrogen bond with protein than protein itself.Two water-protein(hydrophobic residue)hydrogen bonds are highlighted in blue.Water molecule is symbolled by a cyan atom of oxygen bonding with two gray atoms of hydrogen.(e)At nature state,hydrogen bond prefers to form between protein and protein(exactly,between hydrophobic residue and hydrophobic residue).Two protein-protein hydrogen bonds are highlighted by yellow.(f)At hot denatured state,no apparent preference is found in hydrogen bonds between water and protein.(g)Ensemble statistics of the number of hydrogen bonds at cold denatured state,nature state and hot denatured state,respectively.The yellow represents the protein-protein hydrogen bonds and the blue represents the protein-water hydrogen bonds.(h)The water rotational anisotropy v.s.distance between water and protein surface(dotted blue for cold denatured state,solid black for nature state and dashed red for hot denatured state,respectively.)(i)The total water molecule rotational anisotropy calculated by integral for the curve in(h).The data and picture were adapted from Vendruscolo et al.[55].Reprinted with permission from the link https://creativecommons.org/licenses/by/4.0/without change.
In order to obtain an insight into the significance of entropy,the authors analyzed the water molecule rotational anisotropy as a function of distance from protein surface[Fig.9(h)].The reference of zero value represents that the water molecule shows no preference in orientation.Fig.9(i)provides a result of integral for Fig.9(h).The nature state possesses the minimum rotational anisotropy corresponding to minimum water protein hydrogen bonds in Fig.9(g).Compared with nature state,cold denatured state and hot denatured state possess more water molecule rotational anisotropy indicating a more obvious orientation towards protein which can be interpreted as the entropy loss with regard to bulk water.In other words,bulk water possesses the largest entropy at nature state of protein.Accordingly,protein possesses the least entropy with a most stable conformation at nature state in this water-protein interaction system.To be simple,upon decreasing the temperature from 298 K to 272 K or increasing temperature from 298 K to 323 K,protein gains the entropy causing an unstable conformation with expanded structure.However,at the expense of hydrophobic driving force,hot denatured state shows a more stable structure than cold denatured state.
Collectively,hydrophobic driving force plays a significant role in maintaining the compact conformation of protein[3,4,22,59]. It is thought as the most important interaction in protein folding and its temperature dependence broadens applications for biomaterial fields[60].
B.Entropy Convergence in Protein Fold ing
Early in 1980s,Baldwin proposed an entropic convergence concept based on investigation of different liquid hydrocarbons[24].That means at a specific temperature,386 K,various proteins folding amazingly similarly undergoes a thermodynamic process of entropic change of zero.This idea is used to understand hydrophobic contribution to protein folding[61−65].
Huang and Chandler further developed concept of entropy convergence of Baldwin’s on account of length scale dependence mechanism of hydrophobic interaction[22].As shown in Fig.3(b),for small solute,a maximum appears in temperature dependence relation.With increasing solute’s scale length,temperature that corresponds to the maximum would gradually decrease.At much larger scale length,the maximum would vanish.Based on this,Huang and Chandler proposed that entropy convergence was correct for small protein folding but was not able to applied to larger protein folding.
In fact,modeled hard sphere is different from actual proteins.For many globule structure proteins,they are amphiphilic which are more complex than modeled hard sphere. Hydrophobic amino acid residues are buried into inner space under driving force and hydrophilic ones are exposed on the surface.Although hydrophilic residues seem to have more contact with surrounded environment,inner hydrophobic core composed of hydrophobic residues produces a significant effect.Collectively,idea developed by Huang and Chandler achieved a great success in understanding thermodynamic process of protein folding.
C.Protein Folding and Membrane Self-assembly
Massive studies have been paid to energetics of protein folding that determines the three dimensional structure and final morphology of protein.Overall,energy landscape of funnel morphology is a popular model in which there are a lot of possible paths to reach a minimal free energy state from peptides to stable proteins[66,67].At micro level,many secondary bonds such as van der Waals interactions,salt bridges,disulfide bridges and hydrogen bonds take part in modeling the structure of protein and play an essential role in energetics of protein folding.As mentioned above,since hydrophobic interaction is larger than these secondary bonds,it should also undertake a most important mission in energetics of protein folding[60].
Hydrophobic interaction mainly focuses on the burial of hydrophobic residues. Early Kauzmann’s model considered the free energy change of burying a nonpolar side chain calculated by transfer free energy from water to a good solvent like toluene[27].Later research revealed that hydration free energy was proportional to the surface area of hydrophobic residues exposed to water[68,69].Hence,if coefficient k is measured,the free energy change can be calculated by k∆A(∆A is subtraction of surface area between native state and unfolded state).However,after lengthscale dependence theory,calculation method should be corrected again.
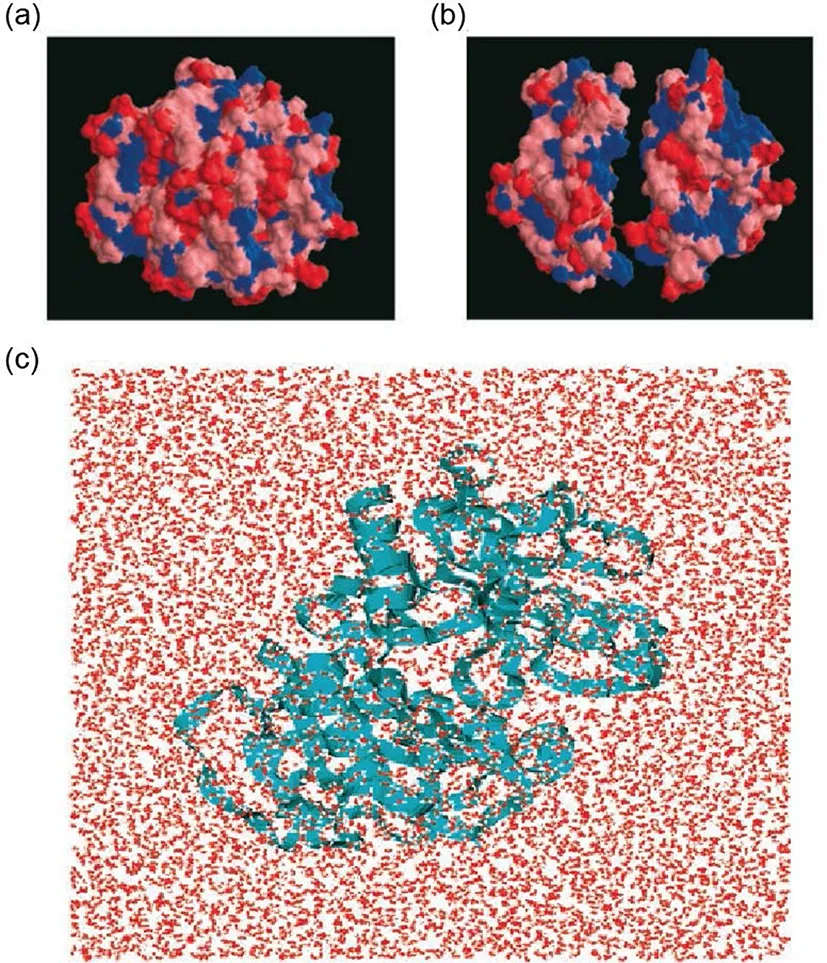
FIG.10.Globular protein,BphC enzyme,with two domains.(a)Surface of folded protein.Blue:hydrophobic residues,Red:strong hydrophilic residues,and Pink:weak hydrophilic residues.(b)Two domains with a distance that show surface of the interface.(c)Solvated protein in water in simulation.The picture was adapted from Ruhong Zhou,Bruce J.Berne et al.Science,2004,305:1605.Reprinted with permission from AAAS.
Since surface is very important in hydrophobic effect,Berne and coworkers mapped the surface of a two domain protein,BphC enzyme,by molecular dynamics simulation in water[Fig.10][70].They found that water molecule cannot be totally excluded from interdomain of BphC.In fact,a 10%∼15%lower density than bulk water was observed in inter-domain even the domain’s separation was very small.This is very different from condition of paraffin-like plates in which water molecule is emptied between plates with small distance[23].Further investigation revealed that protein water attraction played an important role in hydration of protein.Consequently,hydration of protein in actual physiological environment is much more complex than idealized conditions.Hydrophobic force-regulated thermodynamic process of protein folding is influenced by solute-solvent attraction in a certain extent[3,71].
Besides,Cao and coworkers developed a single molecule force spectroscopy assay to study the binding strength between hydrophobin and hydrophobic surface[72].Hydrophobin is one kind of proteins produced by the filamentous fungi.It showed a good attachment to hydrophobic surface rather than hydrophilic one.They first measured the binding strength of individual hydrophobin and various surfaces,including hydrophilic mica,glass and hydrophobic HOPG(Highly Oriented Pyrolytic Graphite),gold.Results of statistical rupture forces suggested that hydrophobin had a stronger binding with hydrophobic surface than hydrophilic one. Next,they measured the binding strength between hydrophobin film and various surfaces.Statistical results of rupture forces were obviously larger than previous values,indicating that hydrophobic assembly on the surface enhanced the binding strength.A reasonable elucidation for this is that the film formed by assembled hydrophobins on the surface enhances hydrophobic interaction.At this length scale,the interfacial free energy is proportional to the surface area of assembled hydrophobin film which is larger than individual state.Besides,interaction among adjacent proteins is not negligible[73,74].Relevant research revealed that hydrophobicity increased the specific protein-protein interaction[75].Therefore,pulling a hydrophobin out of assembled film needs larger force than individual one from the surface.
Hydrophobic interaction was also applied to membrane construction.According to aforementioned introduction,the polymer will collapse into a sphere in water under hydrophobic driving force.Surprisingly,Thordarson and coworkers opened a new path to exert hydrophobic directional aromatic perylene interactions to construct non-spherical polymersomes[76].
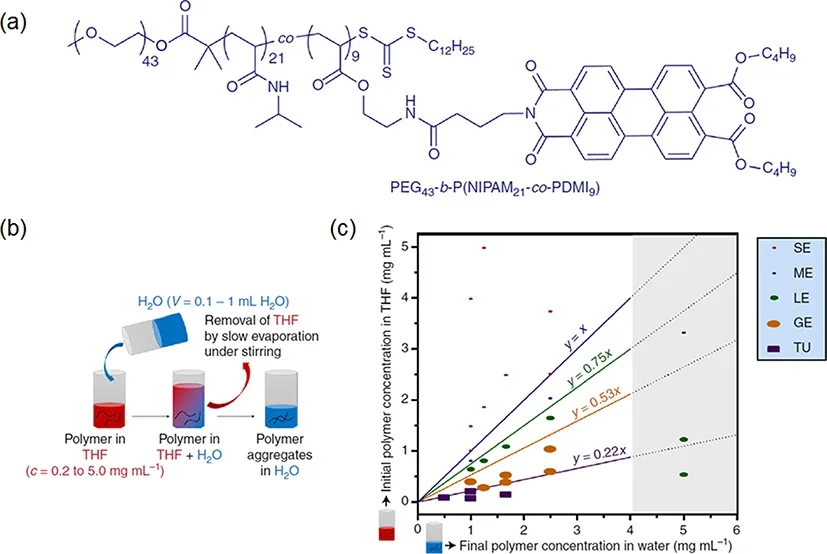
FIG.11.(a)Chemical formula of target terpolymer.The PEG43 moiety provides hydrophilicity and NIPAM 21 moiety is amphiphilic.The most critical moiety of PDMI9 is hydrophobic and provides aromatic interactions(π−π packing)by aggregation in water.(b)Illustration diagram of solvent-switch method for polymer self-assembly.(c)Phase diagram of polymer self-assembly in different THF-water system.GE,giant ellipsoidal polymersomes;ME,medium ellipsoidal polymersomes;SE,small ellipsoidal micelles and TU,tubular polymersomes.Five morphologies transitions are separated by four lines that refer to the different ratio of THF and water.The gray region indicates that phase transition is not obvious owing to the higher polymer concentration in water resulting in a solvation limit of polymer.The data and picture were adapted from Thordarson[76].Reprinted with permission from the link https://creativecommons.org/licenses/by/4.0/without change.
They first synthesized a specific diblock terpolymer, poly (ethylene glycol)-b-poly (Nisopropylacrylamide-co-perylene diester monoimide),(PEG43-b-P(NIPAM21-co-PDMI9))[Fig.11(a)],by reversible addition-fragmentation chain transfer(RAFT)copolymerization.The PEG43moiety is hydrophilic and PDMI9moiety is hydrophobic with a perylene group to give rise to directional aromatic perylene interactions which is the key to the construction of non-spherical polymersomes.Next,they used tetrahydrofuran(THF)to dissolve synthesized polymer and then added water[Fig.11(b)].After that,THF was removed by evaporating for several days to make polymer self-assemble in water.Finally,they obtained the polymersomes with five different morphologies,including tubular polymersomes(TU),giant ellipsoidal polymersomes(GE),large ellipsoidal polymersomes(LE),medium ellipsoidal polymersomes(ME)and small ellipsoidal micelles(SE).Importantly,the final shape of polymersome is decided by the amount of THF and water. Fig.11(c)shows the five shapes transition at different ratio of THF and water.THF of larger amount(corresponding to the lower concentration of polymer)leads to polymersomes with larger size at the constant amount of water.
According to authors’view,thepolymersbeginsto self-assemble immediately upon adding water.The hydrophobic moieties of P(NIPAM21-co-PDMI9)of polymers rearrange and aggregate together to minimize the interfacial free energy due to hydrophobic interactions upon decreasing THF and simultaneously increasing water.However,the hydrophilic moieties of PEG43still maintain extending state upon adding water.Consequently,the local anisotropic characteristic of polymers at molecular level leads to a non-spherical shape of self-assembly.Furthermore,the final higher concentration of polymer in water causes the stronger hydrophobic aggregation that promotes the aromatic interactions(π−π packing)[77,78]which results in a smaller shape of self-assembly[Fig.11(c)].Correspondingly,the initial larger amount concentration of polymer in THF achieves similar results when at final constant concentration of water.
Collectively,the combination of PEG43moieties facing with THF,hydrogen bonding interactions among PEG43and NIPAM21moieties and aromatic interactions among PDMI9 moieties in water forms an anisotropic force that results in a membrane structure of polymersome.This anisotropy is regulated by the ratio of THF and water.Different forces formed at different ratios lead to different shapes.
IV.PERSPECTIVE AND FUTURE
Hydrophobic interaction has appealed great interests of generations of researchers for its significant role in biological and chemical fields.Although length-scale dependence theory achieved great success in unifying different hydration mechanisms,other previous theories did push forward understanding of hydrophobic interaction in history.To a certain extent,length-scale dependence theory is also not a perfect one.With increasing deeper exploration in energetics of protein folding and other relevant issues,length-scale dependence theory gradually seems to be weak to solve such complex ensemble problems for its coarse-grained hard sphere model.However,what can be expected is that a unified theory in energetics of protein folding would be established in the near future.
Putting aside theory itself,there are a lot of applications and developments to deal with that.Burial of hydrophobic residues with a hydrophilic residues coating on the surface forms an energetically favorable structure.Hydrophobic core not only undertakes task of maintaining stable structure,but also interacts with external environment to exert specific functions.Based on this enlightenment,applications for drug carrier can be designed.Hydrophilic coating provides it higher liquidity in blood circulation and hydrophobic core can target with specific receptors.Why not reverse? In a liposoluble environment,hydrophilic core with a hydrophobic coating should be more stable.Facing the barrier of biological membrane,such structure can enter intracellular space in precedence,providing a new idea to transplant target molecule into cell.A much bolder assumption is constructing an amphiphilic surface without inner core.In water,such surface will be newly modified by hydrophobic force.Hydrophobic part sharply contacts with water but hydrophilic part interacts with water stably.The surface will change accordingly to maintain an energetically favorable morphology in environments with different hydrophilicity.As a result,we can screen specific molecules with our desire.
As mentioned above,interface is an important concept.Biological membrane is a kind of interface in nature.Understanding and manipulating interface deserve further investigations[79,80].
Physically,the length-scale dependence theory solves the puzzlement of different hydrophobic hydration mechanisms.Biologically,as the proverb says that one log cannot prop up a tottering building,the length scale dependence theory fails to construct a complete picture of energetics of protein folding and other issues.Extracellular and intercellular environment are much more complex than our imagination.There are many other interactions participating in constructing structure of protein,including salt bridges,disulfide bridges,hydrogen bonds and electrostatic interaction.Recently,some studies revealed that cation-πinteraction played an important role in protein folding and relevant biological issues[81−85].This discovery set emphasis on effect brought by ions.In future studies,hydrophobic force dominated with cation-π interaction assisted combined approach may shed light on energetics of protein folding and other fundamental problems.
We thank Dr.Lei Hai for discussions and Dr.Xue Bin for careful corrections.Bin Xue provided many constructive suggestions to perfect this review work.
