基于正交试验和响应曲面法优化复方金钱草颗粒提取工艺
2020-01-19潘鹏超陈啸飞郭志勇柴逸峰海军军医大学药学院药物分析教研室长海医院肾内科上海200433
潘鹏超,陈啸飞,谌 卫,郭志勇,柴逸峰(海军军医大学:. 药学院药物分析教研室,. 长海医院肾内科,上海200433)
0 引言
复方金钱草颗粒由广金钱草、车前草、光石韦以及玉米须四味中药组成,为成方制剂,收录于《中国药典》(2015年版),该药具有清热利湿、通淋排石的作用,在临床上常用于泌尿系结石、尿路感染证候者[1-3]。现代药理学研究也表明复方金钱草颗粒具有利尿、抑制泌尿系结石形成、抗炎、抗氧化作用,能有效抑制结晶肾损伤,其中,广金钱草作为君药,发挥主要药理作用,车前草和光石韦也起到一定辅助作用[4-7]。经鉴定,复方金钱草颗粒中主要成分包含黄酮类、萜类、生物碱类、酚酸类、甾醇类、多糖类、挥发油等[8-9]。近年来研究表明,其主要活性成分为黄酮类、萜类、酚酸类以及多糖类[10-12]。因其成分复杂,极性强弱各异,目前的中药提取方法主要有煎煮法、加热回流法、半仿生提取法等[13-15]。然而,这些方法对于复方金钱草颗粒中指标性有效成分提取的效率尚无系统研究,因此,目前复方金钱草颗粒尚无通用的提取工艺,有效成分含量未知。
正交试验因其具有较高的科学性,较短的操作时间以及简单的实验步骤被广泛应用于中药材提取工艺优化[16]。响应曲面法通过直观的等高线图和三维立体图比较各因素的相互作用且能通过回归模型预测最佳提取参数,从而在医药食品等领域广泛应用。本研究通过结合正交试验和响应面分析法两种统计学方法,在选定14个复方金钱草颗粒代表性有效成分的基础上,优化得到有效成分含量最大化提取工艺,为后续该药物的药理药效研究奠定基础。
1 仪器与试药
1.1 仪器
200 g摇摆式高速万能粉碎机(温岭市林大机械有限公司);BP121S电子分析天平(德国Sartorius公司);KQ-400KDB型高功率数控超声波清洗器(昆山市超声仪器有限公司);Heraeus Multifuge X1R台式离心机(美国Thermo公司);Agilent1290 液相色谱仪(美国Agilent公司);Agilent 6538 飞行时间质谱仪(美国Agilent公司)。
1.2 试药
分析纯无水乙醇(中国医药集团上海化学试剂公司);质谱纯乙腈(Merck,Germany)、甲酸(Merck,Germany);超纯水(Milli-Q A10超纯水制备系统美国Millipore公司);二甲基亚砜(Merck,Germany);芹菜素对照品、木犀草素对照品、大豆皂苷Ⅰ对照品(上海诗丹德标准技术服务有限公司);复方金钱草颗粒(每袋装10 g)来源于长海医院肾内科。
2 试验方法
2.1 试验因素水平选择
通过前期的文献调研以及对复方金钱草颗粒提取主要因素的考虑,笔者选取超声提取时间(A)、超声温度(B)、乙醇比例(C)作为试验因素并确定了其因素水平表,具体见表1。
2.2 对照品溶液的制备

表1 试验因素水平表
分别精密称取芹菜素、木犀草素、大豆皂苷Ⅰ对照品各5 mg,加一定量的二甲基亚砜(DMSO)溶解,得到浓度均为80 mmol/L的对照品溶液。
2.3 复方金钱草颗粒供试品溶液的制备
取复方金钱草颗粒进行初步粉碎,精密称取2.0 g,置于50 ml锥形瓶中,根据试验方案加入不同比例的乙醇提取液25 ml,称定质量并记录,并按各提取条件进行超声加热回流提取药液,经室温冷却后,再次称定并用提取液补足损失的质量,15 000 r/min离心20 min,取上清液并经0.22 μm滤膜过滤,得各供试品溶液。
2.4 色谱条件
色谱柱:XBridge BEH C18柱(2.1 mm×100 mm,2.5 μm);流动相:A为0.1%甲酸水,B为0.1%甲酸乙腈,梯度洗脱,洗脱条件见表2;进样量为3 μl,流速为0.4 ml/min,柱温为40 ℃。

表2 梯度洗脱条件
2.5 质谱条件
电喷雾离子源(正离子模式);质量扫描范围:50~1 500m/z;干燥气温度: 350 ℃;干燥气流速:11.0 L/min;雾化气压力: 45 psig;毛细管电压: 4 000 V;碎片电压: 120 V。
2.6 标准曲线的制备
2.6.1 芹菜素
取芹菜素对照品溶液,用DMSO溶液稀释得到质量浓度为0.676、1.351、2.027、2.702、6.756、13.512 μg/ml的工作溶液,按“2.4”“2.5”项条件进样测定。以对照品溶液的质量浓度为横坐标,峰面积为纵坐标绘制曲线。
2.6.2 木犀草素
取木犀草素对照品溶液,用DMSO溶液稀释得到质量浓度为0.217、0.716、1.073、1.431、3.578、7.156 μg/ml的工作溶液,按“2.4”“2.5”项条件进样测定。以对照品溶液的质量浓度为横坐标,峰面积为纵坐标绘制曲线。
2.6.3 大豆皂苷Ⅰ
取大豆皂苷Ⅰ对照品溶液,用DMSO溶液稀释得到质量浓度为1.886、7.545、11.317、15.090、22.635、45.270 μg/ml的工作溶液,按“2.4”“2.5”项条件进样测定。以对照品溶液的质量浓度为横坐标,峰面积为纵坐标绘制曲线。
2.7 精密度试验
取浓度为4.054 μg/ml的芹菜素、2.147 μg/ml的木犀草素、18.862 μg/ml的大豆皂苷Ⅰ混合对照品溶液,按“2.4”“2.5”项条件进样,在1 d之内连续进样6次,并且连续进样4 d,测定其芹菜素、木犀草素、大豆皂苷Ⅰ的色谱峰面积并计算其各自的相对标准偏差(RSD),考察其日内精密度和日间精密度。
2.8 重复性试验
取复方金钱草颗粒提取液,平行制备6份,按“2.4”“2.5”项条件进样,连续进样6次,测定其芹菜素、木犀草素、大豆皂苷Ⅰ的色谱峰面积并计算其各自的RSD值,考察其重复性。
2.9 回收率试验
精密量取一定浓度的芹菜素、木犀草素、大豆皂苷Ⅰ混合对照品溶液和已知含量的复方金钱草颗粒提取液各0.5 ml混合,平行制备6份,按“2.4”“2.5”项条件进样,连续进样6次,测定其芹菜素、木犀草素、大豆皂苷Ⅰ的色谱峰面积并计算其各自的含量和RSD值,考察其加样回收率。
2.10 稳定性试验
取复方金钱草颗粒提取液,按“2.4”“2.5”项条件分别在0、6、12、24、48 h进样,测定其芹菜素、木犀草素、大豆皂苷Ⅰ的色谱峰面积并计算各自的RSD值,考察其稳定性。
3 结果
3.1 标准曲线
3.1.1 芹菜素
如图1A所示,回归方程为Y=50 999X+33 373,r=0.999 9,表明芹菜素在0.676~13.512 μg/ml浓度范围内与其峰面积呈良好的线性关系。
3.1.2 木犀草素
如图1B所示,回归方程为Y=45 861X+15 784,r=0.999 9,表明木犀草素在0.215~7.156 μg/ml浓度范围内与其峰面积呈良好的线性关系。
3.1.3 大豆皂苷Ⅰ
如图1C所示,回归方程为Y=19 763X+68 383,r=0.999 9,表明大豆皂苷Ⅰ在1.886 ~45.270 μg/ml浓度范围内与其峰面积呈良好的线性关系。
3.2 精密度试验结果
由表3可知,日内精密度试验中,芹菜素的RSD为0.98%,木犀草素的RSD为1.90%,大豆皂苷Ⅰ的RSD为2.18%,说明这3个化合物的日内精密度良好。日间精密度试验中,芹菜素的RSD为1.53%,木犀草素的RSD为0.87%,大豆皂苷Ⅰ的RSD为1.40%,说明这3个化合物的日间精密度良好。

表3 日内、日间精密度试验结果
3.3 重复性试验结果
平行制备复方金钱草颗粒提取液,重复进样6次,以峰面积计算,结果芹菜素的RSD为3.09%,木犀草素的RSD为2.83%,大豆皂苷Ⅰ的RSD为2.21%。说明复方金钱草颗粒提取液进样测定的重复性良好。
3.4 回收率试验结果
由表4可知,芹菜素的平均回收率为99.58%,其RSD为1.24%;木犀草素的平均回收率为101.07%,其RSD为1.83%;大豆皂苷Ⅰ的平均回收率为100.59%,其RSD为1.22%,说明该测定方法准确性良好。

表4 加样回收率试验结果
3.5 稳定性试验结果
由图2可知,复方金钱草颗粒提取液在0、6、12、24、48 h共进样5次,3个成分的峰面积在48 h内无明显波动,计算得芹菜素、木犀草素、大豆皂苷Ⅰ的RSD分别0.86%、3.83%、1.44%,说明复方金钱草颗粒提取液在48 h内稳定性良好。
3.6 正交试验设计与实验结果
为探讨复方金钱草颗粒最佳提取工艺,笔者采用复方金钱草颗粒中的指标性成分的峰面积之和来代表其提取效果。共选取14种不同的化合物,为代表其合理性,在各味药材中都选取一定数量的化合物,主要为经文献报道的有药理作用的成分以及各味药材中含量相对较高的成分。笔者采用UHPLC-TOF/MS对复方金钱草颗粒提取成分进行鉴定,得到其质谱总离子流图及提取离子流图(图3),其化合物详细信息见表5。
采用L9(33)正交试验表划分各个试验的因素水平,其具体的试验方案和结果分析见表6。根据表6的试验结果分析,通过比较K值,得到其提取最优参数水平为A1、B2、C2,通过比较极差R值,可以看到因素A的极差最大,其次为C、B,即超声提取时间对复方金钱草颗粒的提取效果影响最大,其次是乙醇比例,最后是超声温度。结果表明,适宜超声提取时间和温度能促进化合物的溶出,但当超声提取时间过长、温度过高时,可能会破坏物质的结构,使得溶出的化合物含量反而降低;适宜的乙醇比例可以使复方金钱草颗粒中的水溶性和脂溶性成分都能最大限度的溶出。

表5 复方金钱草指标性有效成分列表

表6 正交试验结果与分析
进一步采用IBM SPSS Statistics 19软件对正交试验结果进行方差分析,各因素对复方金钱草颗粒的提取效果由F值和P值进行检验,具体结果见表7。
对表7结果进行分析,影响复方金钱草颗粒提取效果的因素中,A>C>B,这与上述根据表6的极差得到的结果一致。显著性分析发现,因素A和因素C的P值<0.05,差异显著,而因素B>0.05,其差异无统计学意义。

表7 方差分析表
3.7 响应曲面试验设计与实验结果
采用软件Design-Expert 10,以超声提取时间、超声温度、乙醇比例为自变量,指标性化合物峰面积之和为响应值设计试验。共设17组试验点,其中,分析点12个,零点5个,零点代表重复实验,常用于估算试验误差。本试验方案及结果见表8。
将表8的试验结果输入Design-Expert 10软件进行综合分析,可以得到复方金钱草颗粒指标性成分峰面积之和与各因素变量的回归方程模型:
通过对表9的数据进行方差分析,可以看到该模型的P<0.01,r=0.960,失拟项显著,说明该模型虽存在试验误差,但模型拟合度较好,能够用来预测复方金钱草提取效果与各因素变量的关系。同时,各因素显著性分析显示,超声提取时间(A)以及乙醇比例(C)的P<0.05,说明它们对复方金钱草颗粒的提取效果作用明显,而超声温度对其提取效果影响不显著,这与前面的正交试验结果相一致。

表8 响应曲面试验方案及结果
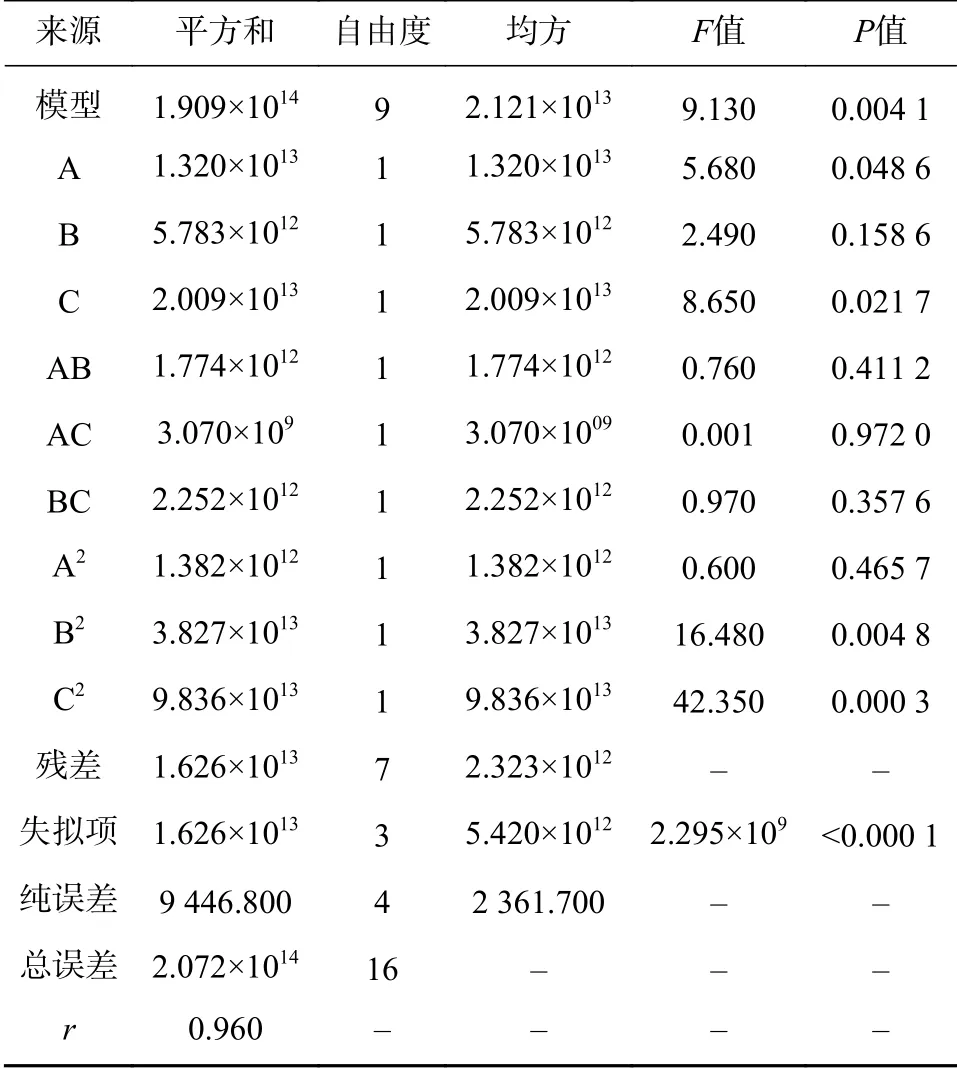
表9 响应曲面二次回归模型的方差分析
3.8 响应曲面各因素间的交互分析
利用Design-Expert 10软件可以得到其等高线图和3D响应曲面图,如图4所示。其中,等高线的形状反映两者交互作用的强弱,圆形代表交互作用弱,椭圆形代表交互作用强。观察发现,A图显示的超声提取时间和乙醇比例交互作用最强,B图显示的超声提取时间和超声温度交互作用次之,C图显示的超声温度和乙醇比例交互作用最弱。
3.9 复方金钱草颗粒最优提取参数预测及验证
使用回归模型进行最优提取参数预测,得到当超声提取时间为10 min,超声温度为55.12 ℃,乙醇比例为54.56%时,得到的指标性峰面积之和为77 471 863.44。结合实际操作,选用超声提取时间10 min,超声温度55 ℃,乙醇比例55%进行验证,经3次平行试验,得到其平均值为77 978 123.33,RSD为1.91%,与模型的预测值基本一致,表明该预测模型的稳定性强、适用性良好。
4 结论
本研究的评价指标为14个指标性有效成分的峰面积之和,以乙醇为提取溶剂,采用正交试验和响应曲面法进行复方金钱草颗粒提取工艺的优化,正交试验结果显示,其最佳提取工艺参数为超声提取时间10 min,超声温度60 ℃,乙醇比例50%;响应曲面法则通过回归模型对复方金钱草颗粒提取工艺参数进行预测,得到其预测的最佳参数为:超声提取时间10 min,超声温度55 ℃,乙醇比例55%,通过两种试验方法得到的参数基本一致,且通过响应曲面法预测得到的峰面积之和更大。综上所述,我们选择工艺参数为:超声提取时间10 min,超声温度55 ℃,乙醇比例55%,并在此提取条件下进行验证,其UHPLC-TOF/MS图谱中峰面积之和平均值为77 978 123.33,说明该模型稳定可靠,可以用于复方金钱草颗粒的提取。
由于前期采用水提法提取复方金钱草颗粒,导致水溶性小的成分没有被完全提取出来,故而改进方法,采用一定比例的乙醇溶液为提取溶剂,并且优化了最佳提取时间和温度,使得其指标性有效成分都能被有效提取,并为后续复方金钱草的药效和药理研究提供可靠的方法依据。
