Comparative transcriptome analyses reveal candidate genes regulating wood quality in Japanese larch(Larix kaempferi)
2020-01-18ShaeHeYunhuiXieXiaomeiSunShougongZhang
Shae He·Yunhui Xie·Xiaomei Sun·Shougong Zhang
Abstract We studied the molecular mechanism of the quality traits of wood formation in larch.We used the immature latewood cells of two Japanese larch(Larix kaempferi)clones with significant differences in density and in microfibrillar angle(MFA)as materials to analyze their gene expression profiles.A total of 1735 differentially expressed genes were detected in immature latewood cells of the two clones,among which,971 were up-regulated and 764 were down-regulated.Digital gene expression profiling analysis revealed that genes encoding transcription factor members NAC66 and R2R3-MYB4,microtubule-associated protein,actin-related protein,cell wall protein members,arabinogalactan protein,Fasciclin-like arabinogalactan protein and glycine-rich protein, and several cell-wall-synthesis genes affected wood density and MFA by regulating latewood formation at transcriptional level.Our study results represent a basis for selection of quality traits and genetic improvement of larch wood.
Keywords Japanese larch·Wood formation·Transcription regulation·Wood property·Digital gene expression profiling
Introduction
Forest breeders aim to genetically improve the mechanical properties of wood and fiber by selecting traits that can yield high-quality wood products.Wood density reflects the accumulation of cell wall components while microfibrillar angle(MFA)reflects the structure of the fiber in the cell wall(Evans and Ilic 2001).These two traits influence the mechanical properties of wood.However,wood property traits are complex and are regulated through the expression of multiple genes at the transcription level(Hertzberg et al.2001;Schrader et al.2004).Analysis of wood-formation transcription regulation is the basis for breeding improvement through selection of quality traits.Researchers have increasingly focused on isolation and identification of wood-related genes associated with transcription regulation and quality traits by using genetic engineering and gene localization and transcription techniques.
Wood formation results from the development of secondary xylem,a fundamental biological mechanism of tree growth.Given the long growth cycle of trees and the conservative nature of gene expression in the secondary growth of vascular plants,most studies have been conducted through the secondary-wall synthesis of model herbs,such as the zinnia(Pesquet et al.2005),Arabidopsis thaliana(Zhong et al.2010b),and model tree species such as poplar(Zhong et al.2010a,2013;Li et al.2019).Several key genes involved in secondary-wall synthesis have been isolated and identified,and the transcription regulation network of poplar secondary wall formation has been inferred(Ye and Zhong 2015).The regulatory network includes multiple transcription factors and cell-wall-synthesis genes(lignin,cellulose,hemicellulose,and others),as well as cell wall-related proteins,cytoskeleton related proteins,and other members(Ye and Zhong 2015).The expression of these regulatory factors and genes determines the composition,structure of cell wall,and interaction between cells,and it influences the quality traits of wood(Ye and Zhong 2015;Camargo et al.2019).
Several genes related to wood density and MFA have been reported.The cytoskeleton gene β-tubulin in eucalyptus can affect the localization of cellulose microfilaments in cell walls(Spokevicius et al.2007),and the arabic galactose proteins FLA11 and FLA12 can alter the deposition of cellulose microfilaments and the accumulation of cell wall components(MacMillan et al.2010).The polymorphism of the lignin synthesis gene CCR in eucalyptus is related to the microfibrillar angle(MFA)(Wightman and Turner 2008);the allelic variation of the lignin synthesisrelated genes,namely,PAL1,PCBER2,and COMT,in radiata pine is related to wood density and hardness(Dillon et al.2010).Secondary wall polysaccharide biosynthesis genes are associated with wood density(Mizrachi et al.2017).Although these studies have reported genes related to wood density and MFA,little is known at the transcriptional level.
High-throughput sequencing technology is used to study the formation of wood and analyze the expression level of the entire transcription of different materials.Using this technology,Li et al.(2010)found that under natural conditions,the transcriptome of xylem is consistent with the variation of the MFA and mechanical properties of radiata pine wood,especially during latewood formation.Li et al.(2011)reported that the expression of a cytokinin binding protein is abundant in wood with a lower fiber filament angle.By comparing the gene expression of compression wood and normal wood,Yuan et al.(2010)found that the 1-aminocyclopropane-1-carboxylate(ACC)oxidase gene,a key enzyme in ethylene synthesis,significantly increased in the expression level of compressed wood.By comparing the gene expression of poplar tension wood and normal wood,Chen et al.(2015)found that Pt-LIM1/LIM2 and four MYB(Pt-MYB090/156/721/167)transcription factors were involved in lignin-synthesis regulation,and transcription factors such as VNI2,VND7,and MYB103,are involved in the regulation of cellulose-synthesis pathways.
Coniferous species of northern forests are economically important because they provide wood products.However,the study of the transcription regulation of coniferous wood formation lags behind that of broad-leaved tree species,such as poplar(Camargo et al.2019).Larch(Larix spp.)is a commercially exploited coniferous tree species of northern China.Study of larch wood formation is of both theoretical and practical importance. We studied two Japanese larch(Larix kaempferi)clones with significant differences in wood density and MFA to analyze the gene expression patterns of immature latewood cells at transcription level by using digital gene expression profiling(DGE).We aimed to identify candidate genes involved in the wood formation of larch,especially in the regulation of MFA and wood density,and to develop a theoretical basis for the selection of quality traits and genetic improvement of larch wood.
Materials and methods
Experimental materials and total RNA extraction
The Japanese larch used in the experiment was acquired from the clonal test forest of the Wu Ma Temple Forest Farms(34°14′N,112°07′E)at 1400-1600 m a.s.l.,Son Town, Henan province, China. The age of trees was 16 years.The clonal trial was set in a randomized complete block design consisting of four replication blocks of 78 different clones.In each replication block,four ramets of each clone were originally planted at a spacing of 2 m×2 m.Following(Chen et al.2018),two clones that showed significant differences in density and in MFA,namely,clone 1(C1)and clone(C2)were used as materials,and three trees of each clone were treated as biological repetitions(Table 1).Since wood traits are mainlyaffected by latewood(Chen et al.2018),immature latewood cells were sampled on 3 September 2014 and prepared as follows:the bark and phloem were peeled offat breast height;the immature xylem was scraped with a blade;xylem samples were wrapped in tinfoil paper,placed quickly in liquid nitrogen to freeze,and stored at-80°C.Total RNA was extracted using an improved CTAB method(Pavy et al.2008),and RNA integrity was detected by a 2100 Bioanalyzer(Agilent Technologies).
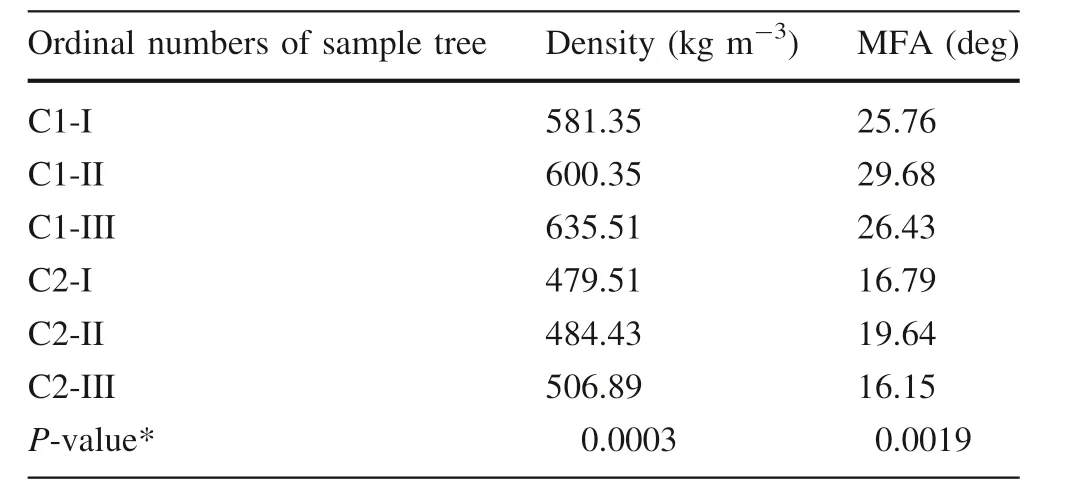
Table 1 Wood property traits of two Japanese larch clones
Construction of differential gene expression library
DGE was completed by the BioMaker Biotech Co.,Ltd.The concrete steps were as follows:(1)After extracting the sample total RNA,mRNA was separated from total RNA with a magnetic bead with oligo(dT)and interrupted into small fragments by adding lysate.(2)The first chain of cDNA was synthesized by random primer reverse transcription with the interrupted small fragment mRNA.(3)Buffer was added,and the second chain of cDNA was synthesized by dNTPs,RNase H,and DNA polymerase I.(4)cDNA was purified by Qiaquick PCR purification kit(Qiagen)and added to the buffer EB to elute.After elution,the samples were modified at the end and connected with the 3′end and the A connection sequencing joint.Then,agarose gel electrophoresis was used for fragment size selection.Finally,we amplified the samples by PCR and sequenced the sequencing library with Illumina HISEQTM 2500.
Analysis of gene expression
After evaluating the data of the original reads obtained by sequencing,we removed the artificial sequence including sequencing primers and adapters and filtering low-quality data,then cleaned the data for the subsequent analysis.Using bowtie,we compared the clean data with the existing Japanese larch transcriptome (accession number:SRR602316).According to the comparison information,the expression level was estimated by RSEM.Finally,the reads per kilobase per million mapped reads(RPKM)value was used to reflect the expression abundance of the corresponding unigenes.
Differentially expressed genes(DEGs)screening and functional annotation
The DEGs were screened with DESeq software. The domain value of P value in multiple detection analysis was determined by controlling the False Discovery Rate(FDR).Finally,the results FDR <0.01 and fold change ≥4 were defined as significant differences in gene expression.Using BLAST software,we compared the DEG sequences with NCBI nonredundant protein database (Nr), Swiss-Prot protein database,gene ontology(GO),clusters of orthologous groups(COG),and Kyoto Encyclopedia of Genes and Genomes Pathway (KEGG) databases, to derive the annotation information of DEGs.
Validation of DGE data by real-time quantitative reverse transcription polymerase chain reaction PCR(qRT-PCR)
Gene-specific primers used for real-time RT-PCR(qRTPCR)were designed online(http://www.ncbi.nih.gov/tools/primer-blast/)(Table S1).Total RNA was a portion of that used for DGE experiments.First-strand cDNA was synthesized using the EasyScript One-Step gDNA Removal and cDNA Synthesis SuperMix kit(TransGen Biotech,Beijing,China).qRT-PCR reactions were conducted in triplicates in 20 μL volumes containing 5 μL diluted cDNA,300 nM of each primer,and 10 μL of the Thunderbird SYBR Green PCR Master Mix.The qRT-PCR program was running on a real-time PCR system(LightCycler®480II,Roche,Tokyo,Japan)set to the following conditions:95°C for 2 min,40 cycles at 95°C for 15 s,60°C for 15 s,and 72°C for 15 s.Dissociation curves were performed at 95°C for 15 s,60°C for 15 s,and 95°C for 15 s for the detection of the specificity of the amplification products.Relative expression level of each gene was calculated using the 2-△△Ctmethod and the elongation factor 1(EF1;accession number:JR146298)was used as the internal control gene.
Results
Gene expression of immature latewood cells
To explain the difference of wood density and MFA between C1 and C2 from the transcription level,we conducted DGEs of the two clones during latewood formation.We then annotated the obtained data with the existing transcriptome of Japanese larch (accession numbers:SRR602316).Totals of 11.4-12.7 million clean reads were obtained for each sample with Q30 percentage over 92%and GC percentage between 44.29 and 46.03%(Table 2).Over 76%clean reads were mapped to the reference transcriptome of Japanese larch,at least 57%of which were uniquely mapped(Table 2).
Differentially expressed genes(DEGs)and annotation
By filtering with FDR ≤0.01 and fold change ≥4,1735 genes in C1 had changed significantly compared with thosein the immature xylem library of C2.Among these,971 gene expression levels were up-regulated and 764 gene expression levels were down-regulated compared with those in C2.Thus,the genes had different transcription expression patterns during the latewood formation of the two clones.
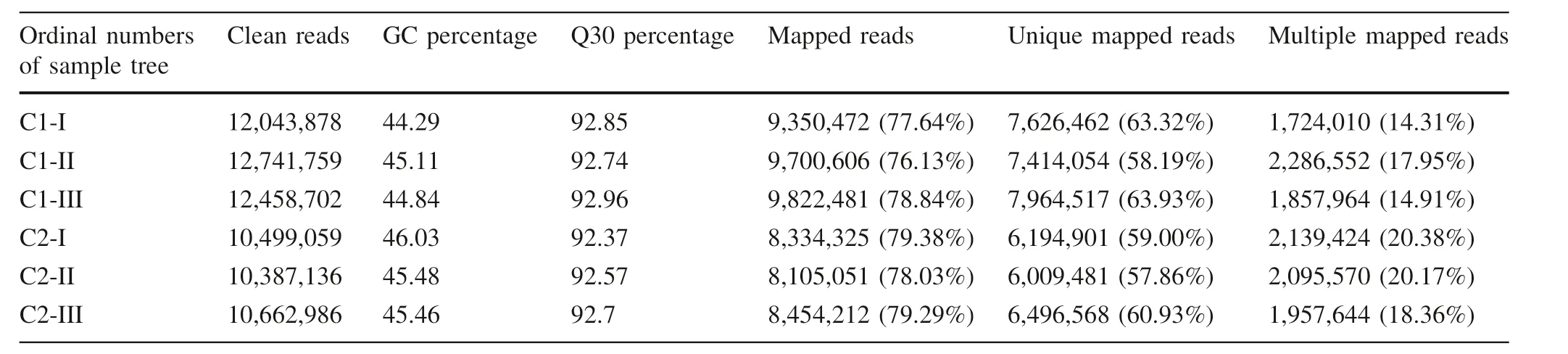
Table 2 Information of clean reads obtained by digital gene expression profiling(DGE)in the immature latewood cells of two Japanese larch clones with different wood property traits
Using BLAST software,in 1735 DEGs,1340 and 904 sequences obtained functional annotations in Nr database and Swiss-Prot,respectively(Table 3).Furthermore,815 sequences were categorized into 42 functional groups(Fig.1).In the three main GO classification groups(cell components, molecular functions, and biological processes),the cell components,catalytic activity,and metabolic processes accounted for the main part, whereas organelles,binding functions,and cellular process classes also accounted for a higher proportion(Fig.1).A total of 359 sequences had COG classification(Table 3).In 22 COG classifications, the sequences expressed as ‘‘just general functional predictions''were most abundant(81%),followed by ‘‘carbohydrate transport and metabolism''(46%),‘‘post-translation modification,protein folding,and molecular companion'' (41%), ‘biosynthesis of cell walls/cell membranes''(40%),and‘‘transcription''(38%).Moreover,the DEG with 232(13%)sequences was annotated in KEGG(Table 3).Some DEGs had‘‘no match''or‘‘unknown functional proteins''that might tree specific genes and play an important role in latewood formation.Among the DEGs that were annotated,some NAC transcription factors, R2R3-MYB transcription factors,cytoskeleton-related proteins,cell wall-structure proteins,cellulose and lignin synthesis genes were included,and the difference of expression levels of these genes in two clones were more than fourfold(Fig.2,Table S2).
The accuracy of DGE data
To further verify the accuracy of DGE data,10 DEGs were selected randomly for qRT-PCR analysis(Table S1).The expression levels of genes encoding pectinesterase,transcription inhibitor KAN,Leucine-rich repeat receptor-like protein kinase PXL2, WAT1-related protein, WARK transcription factor,and two unknown functional genes in C2 were significantly higher than those in C1(Fig.3).The expression levels of the three other genes,namely,laccase-17,R2R3-Myb4,and serine carboxypeptidase encoding gene,in C2 were lower than those in C1.The qRT-PCR results proved consistent with the DGE results(Fig.3),further demonstrating the reliability of the sequencing results.
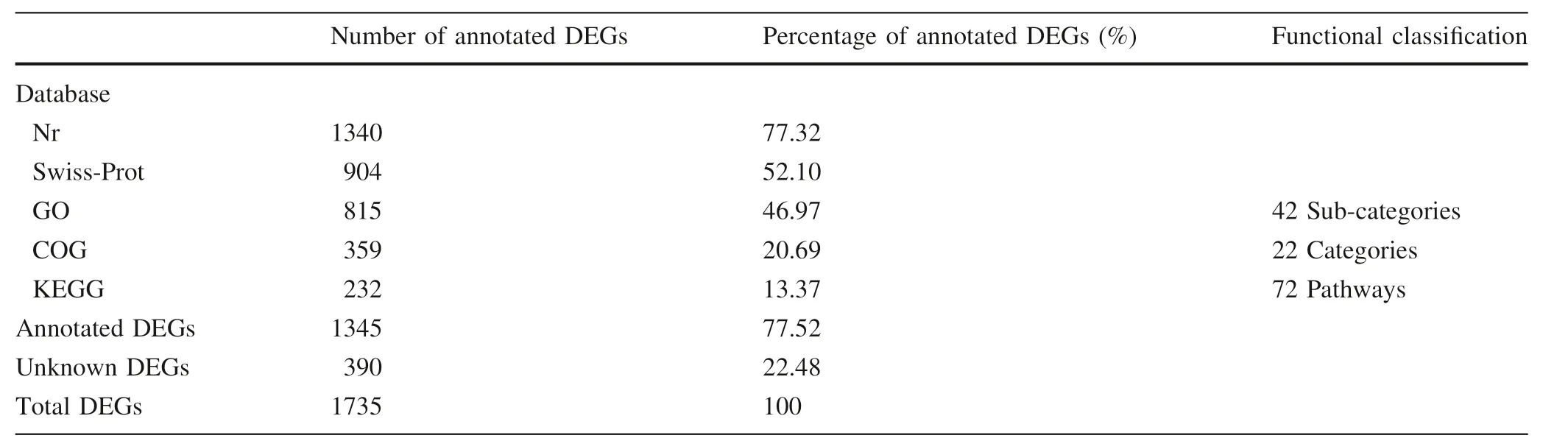
Table 3 Annotation information of differentially expressed genes(DEGs)between the two Japanese larch clones with different wood property traits
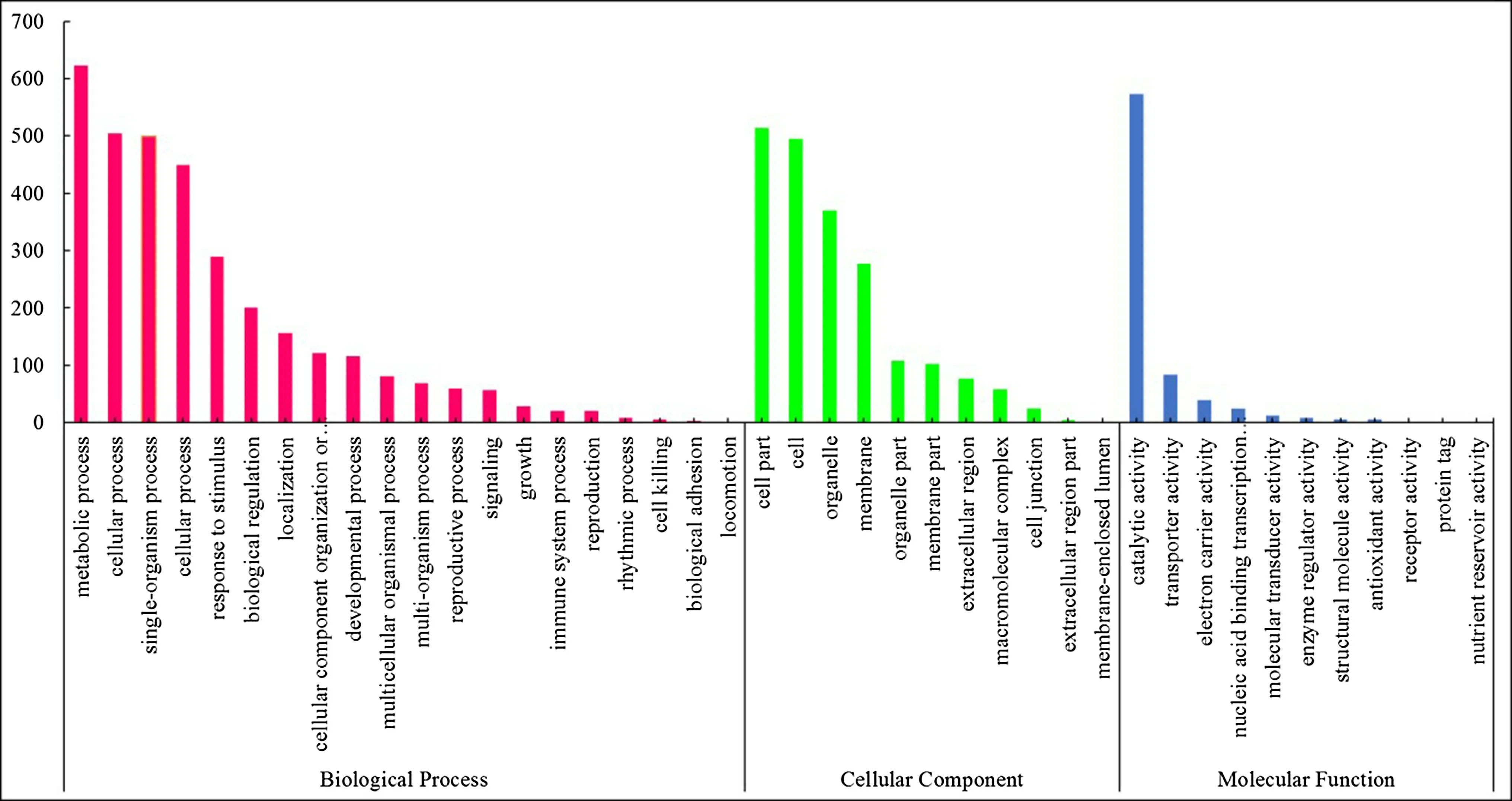
Fig.1 Gene ontology(GO)classifications of differentially expressed genes(DEGs)between two Japanese larch clones with different wood property traits
Discussion
Transcription factor
Some family members of transcription factors(e.g.,NAC,MYB,bHLH,WRKY,Thrihelix,Hdzip III,LBD and zinc finger protein)are involved in cell wall synthesis(Izhaki and Bowman 2007;Legay et al.2010;Ohashi-lto et al.2010;Chai et al.2014).In particular,some members of the NAC and MYB families control the secondary-wall-synthesis regulatory network so they can directly regulate the expression of cell-wall-synthesis genes.They can also be used for other transcription factors to indirectly regulate the synthesis of secondary walls(McCarthy et al.2011;Kim et al.2013;Nakano et al.2015).The control network of secondary-wall synthesis in coniferous species has remained unknown. Nevertheless, studies in pine and spruce have shown that some members of the R2R3-MYB family are essential regulatory factors for lignin deposition and secondary-wall formation,and they are activated and combined with AC(ACCTACC)components by NAC(NAM/ATAF/CUC) transcription factors by interacting with other R2R3-MYB members to affect gene expression(Bedon et al.2007;Zhong et al.2008).The homologous genes of R2R3-Myb4 in A. thaliana and poplar were located in the key position of secondary-wall-synthesis control network,directly activating the genes that synthesize cellulose,lignin,and hemicellulose,and regulate the expression of other transcription factors(Ye and Zhong 2015).In this study,three sequences of encoded NAC transcription factors(Fig.2,Table S2)were down-regulated in C2.In addition,multiple R2R3-MYB transcription factor members were expressed differently in two clones.Among these members,R2R3-Myb4(Fig.2,Table S2,riben_isotig14723)was almost not expressed in C2.Other MYB functions were obscure,but most of the MYBs that had different expression levels were consistent with the expression patterns of R2R3-Myb4,both of which were down-regulated in C2(Fig.2,Table S2).These transcription factor genes were expressed differently in different materials of Japanese larch,illustrating that their transcription regulation affects the quality of larch wood.
Cell-wall-synthesis gene and cell wall-related protein
Studies of radiata pine have shown that the expression of primary and secondary-wall synthetic genes is related to wood MFA;genes involved in secondary-wall synthesis such as PrCesA1,3,7,11,C4H,C3H,CAD,PCBER,4CL,CCoAOMT,chitinase-like,SAMS,PPBG,AGP4,GRP2,and PRP,are preferred in wood with extreme hardness and low MFA(Li et al.2011).In this study,the genes involved in secondary-wall synthesis(CesA4,7,8,4CL,CCoAOMT,COMT,C4H,and Laccase)expressed differently in two clones(Fig.2,Table S2).These genes were not,however,preferred in the C2 with low MFA,possibly because these genes have different family members in the plant,and different members perform different functions.For example,the 4-coumarate coenzyme ligase(4CL)can participate in secondary-wall synthesis,and it is also a key gene for metabolic pathways such as flavonoids and anthocyanins(Shi et al.2010).Thus,the complexity of the regulation of wood quality traits needs to be studied further.

Fig.2 Heatmap of 72 differentially expressed genes(DEGs)encode transcription factors,cytoskeleton-related proteins,cell wall-structure proteins and cell wall biosynthesis enzymes in immature latewood cells of the two Japanese larch clones with different wood property traits.The bar indicates the normalized RPKM value of each gene(log2RPKM).Red and blue indicate over-and underrepresented genes,respectively;black indicate unexpressed genes

Fig.3 Confirmation of the expression levels of 10 differentially expressed genes(DEGs)by qRT-PCR.The relative expression level of each gene measured by the qRT-PCR was normalized to the expression level of the internal control gene(elongation factor 1)in each clone.Fold difference in gene expression level was calculated by dividing the relative expression level in the clone1(C1)by which in the clone2(C2)
CesAs and SuSy might relate to the wood density,as the expression of secondary-wall synthesis-related CesAs in compressed wood of radiata pine(Li et al.2009),maritime pine(Villalobos et al.2012)and tension wood of eucalyptus(Lu et al.2008)and poplar(Chen et al.2015)were up-regulated,and the expression level of SuSy was also upregulated in the compressed wood of maritime pine(Villalobos et al.2012)and radiata pine(Li et al.2013).In addition, genetic function studies have shown that expression of the SuSy gene in poplar leads to increased cellulose content,cell wall thickness,and wood density(Coleman et al. 2009). The strong inhibition of SUS activity reduced the wood density of the transgenic poplar and was accompanied by a decrease in lignin,hemicellulose,and cellulose content(Gerber et al.2014).In this study,C1 and C2 had different characteristics of MFA and wood density in phenotype.In gene expression,CesA1,4,7,8,9,and 9 of 10 and sucrose synthase gene(SuSy)copies were all up-regulated in C1(Fig.2,Table S2).The upregulated expression of these genes might affect MFA,and it might promote the synthesis of cellulose and lignin,possibly one of the reasons why C1 had greater density.
Cell wall structural proteins are associated with MFA.For example, poplar proline-rich protein regulated the expression of genes related to MFA and secondary-wall synthesis,and finally changed the MFA value of wood(Li et al.2019).In addition to changing the MFA of wood,the FLAs in transgenic eucalyptus and A.thaliana can also alter the content of cellulose,galactose,and other cell-wall substrates and affect the compressive/tensile properties of wood(MacMillan et al.2010).In this study,cell-wallstructure protein-coding genes such as Fasciclin-like arabinogalactan protein 18, arabinogalactan protein and glycine-rich protein were expressed differently in two clones(Fig.2,Table S2),suggesting their relationship with the quality of larch wood.
Cytoskeleton-related genes
The cytoskeleton consists of microtubules,microfilaments,and intermediate fibers.The cortical microtubules guide the sedimentary direction of microfibrillar in the S2 layer of the secondary wall and affect the MFA(Spokevicius et al. 2007). Microtubule-associated proteins (MAPs)control the kinetic stability of microtubules and the interaction of intermediate fibers(Mao et al.2005),and A.thaliana AtMAP65-8 was involved in xylem development(Struk and Dhonukshe 2014).In the present study,several genes such as MAP65-1,MAP65-8,two members of the RP/EB family(1B and 1C),TORTIFOLIA1,and five Kinesin proteins,encoded with microtubule-associated proteins were significantly different in two clones(Fig.2,Table S2).With the exception of RP/EB-1B,other microtubule-associated protein-coding genes showed lower levels of expression in C2(Low MAF).Hence,the differential expression of these genes may be related to the difference of wood material in the two clones,especially the difference of MFA.
Actin is a component of microfilaments.According to genomic information analysis,poplar contains eight ACT family members,and the analysis of microarray and qRTPCR expression shows that they are preferred in wood formation,indicating that Actin plays a role in wood formation(Zhang et al.2010).Actin can guide the distribution of cellulose synthase complexes during secondary-wall synthesis(Wightman and Turner 2008).In this study,the expression of two actin-related protein-coding genes was upgraded in C1(Fig.2,Table S2),this finding is consistent with the results of poplar studies(Zhang et al.2010),suggesting their relationship with wood formation.However,their influence on the quality traits of wood has yet to be fully studied.
Conclusions
By comparing the pattern of xylem gene expression during the latewood formation of Japanese larch clones with different wood properties,we found that the important transcription factors (NAC66, R2R3-Myb4) had higher expression levels in the high MFA and high density clone(C1).The difference was more than fourfold.Thus,these transcription factors are likely to be the core regulatory factors for the latewood formation in Japanese larch.In addition,the expression levels of encoded genes,including cytoskeleton genes(MAP protein,ACTIN-associated protein),cell wall synthesis-related genes(CesA,Susy,4CL,C4H, COMT, Laccase), and cell wall-related proteins(AGPs, FLAs, glycine-rich proteins) changed greatly.Furthermore,many genes with unclear functions were also expressed,possibly leading to the differences in the main components and structures of wood cells.These genes might have played important roles in the formation of Japanese larch latewood.This study provides resources for the future development of genetic breeding of Japanese larch,especially for molecular marker-assisted breeding and forest genetic improvement directed at improving the MFA and mechanical properties.
Open AccessThis article is distributed under the terms of the Creative Commons Attribution 4.0 International License(http://crea tivecommons.org/licenses/by/4.0/),which permits unrestricted use,distribution,and reproduction in any medium,provided you give appropriate credit to the original author(s)and the source,provide a link to the Creative Commons license,and indicate if changes were made.
Author's contributionsSGZ,YHX and XMS designed the study;SEH performed the experiments,SEH and YHX analyzed the data;SGZ and SEH wrote the article.
杂志排行

Journal of Forestry Research的其它文章
- Protective and defensive roles of non-glandular trichomes against multiple stresses: structure-function coordination
- Assessment of early survival and growth of planted Scots pine(Pinus sylvestris)seedlings under extreme continental climate conditions of northern Mongolia
- Influencing in vitro clonal propagation of Chonemorpha fragrans(moon)Alston by culture media strength,plant growth regulators,carbon source and photo periodic incubation
- Variation analysis of growth traits of four poplar clones under different water and fertilizer management
- Nodule study in Albizia chinensis in relation to nitrogen metabolism,morphology and biomass
- Comparative study on the mRNA expression of Pinus massoniana infected by Bursaphelenchus xylophilus