误诊喉肿物的复发性多软骨炎1例及文献复习
2019-12-27连媛媛,苗贵申,韩晓华,黄沂传,刘婷婷,徐禛,黄天桥,张念凯
连媛媛,苗贵申,韩晓华,黄沂传,刘婷婷,徐禛,黄天桥,张念凯
关键词:复发性多软骨炎;呼吸困难;喉肿物
中图分类号:R681.3 文献标识码:B DOI:10.3969/j.issn.1006-1959.2019.22.068
文章编号:1006-1959(2019)22-0190-03
1临床资料
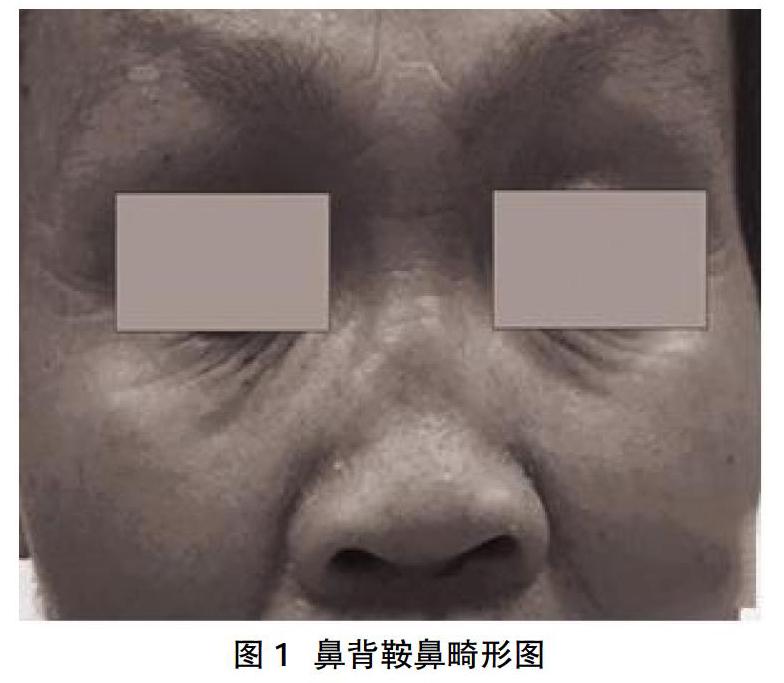
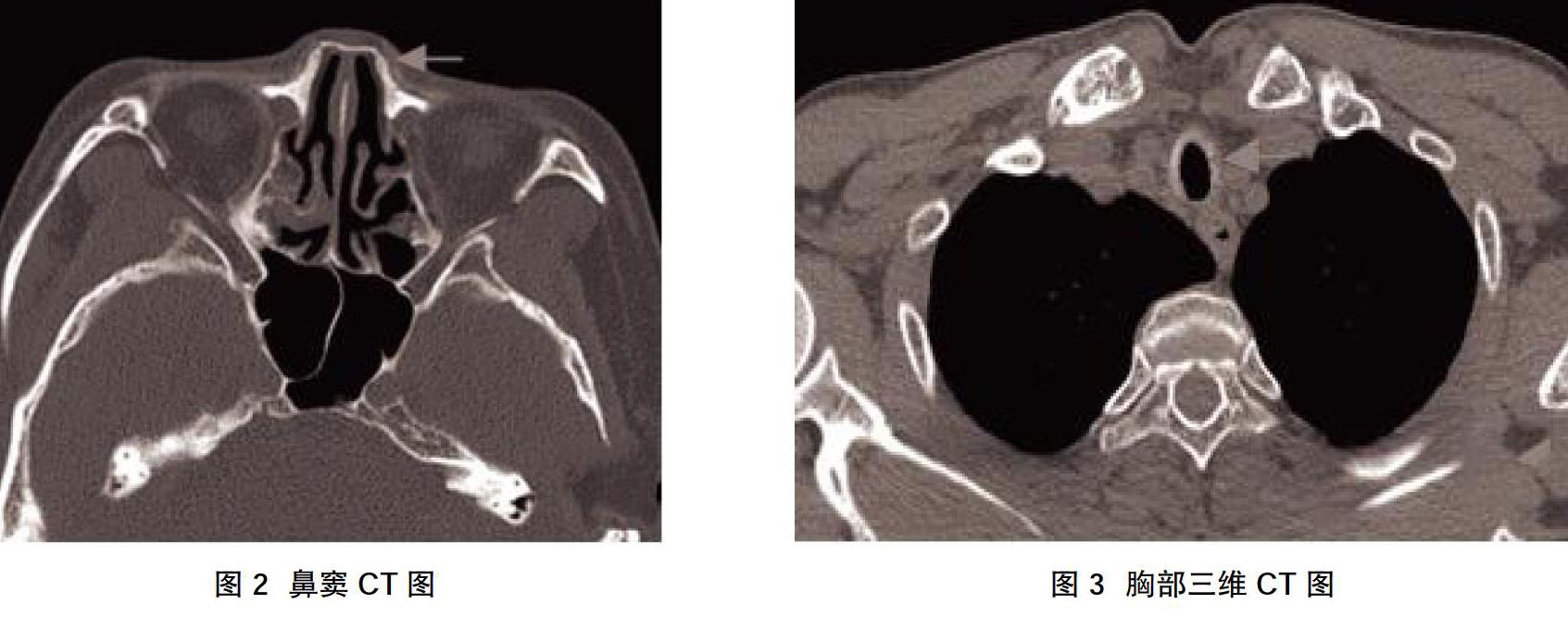
仕某,男,51岁,因“呼吸困难2年,加重半年”于2018年1月18日就诊于青岛大学附属医院。患者6年前因“声嘶1年余,加重伴呼吸困难半年”就诊于我院,行电子喉镜检查示:左侧披裂向中线移位,喉室饱满,声带固定,未见新生物;颈部增强CT示:左侧喉旁间隙肿物,边界欠清。初步诊断:喉肿物,并于全麻下行支撑喉镜下喉肿物活检+喉裂开喉肿物活检+气管切开术+气管成形术,病理结果示:(左室带旁)增生的纤维组织伴纤维化、轻度慢性炎,部分区域软骨岛形成(软骨化生?)及小片状骨样组织,未见恶性特征。术后恢复情况良好,定期门诊复查,1年后顺利拔除气管套管。2年前患者“上呼吸道感染”后出现呼吸困难,表现为活动后加重,睡眠时可平卧,伴有咳嗽、偶有白痰,于当地医院就诊,诊断为“气管炎”,予“抗生素”治疗,疗效欠佳,症状反复发作,未予重视。半年前呼吸困难加重,睡眠时无法平卧,伴有咳嗽,偶有白痰,于当地医院抗炎治疗,效果欠佳。后于我院急诊就诊,考虑喉梗阻(Ⅲ度),急诊入院。患者自发病以来精神好,饮食可,睡眠欠佳,大小便无明显异常,体重无明显异常变化。既往反复上呼吸道感染史7年,4~5次/年,症状主要为鼻塞、流涕、咳嗽;结膜炎病史6年余,胃溃疡病史1年;否认吸烟史,饮酒史30年,偶有饮酒,戒酒6年。入院查体:T:37℃,P:80次/min,R:20次/min,BP:120/70 mmHg。三凹征(+),双肺听诊均可闻及哮鸣音。耳廓无红肿,无畸形,外耳道通畅,鼻背塌陷(图1),无红肿。鼻前庭皮肤无红肿。鼻中隔居中,无血肿。会厌无卷曲,黏膜无红肿,声门裂狭窄,左侧半喉似固定,右侧声带外展受限,声门下狭窄。初步诊断:①喉梗阻(Ⅲ度);②喉术后;③复发性多软骨炎?入院后给予Ⅰ级护理,持续吸氧、心电监护、持续血氧饱和度监测;甲强龙40 mg静脉滴注,普米克令舒、博利康尼雾化吸入,沐舒坦静滴化痰;完善血液相关检查及胸部三维CT、颈部CT、鼻窦CT;请风湿免疫科、呼吸内科会诊,并因呼吸困难急性加重,给予床旁行紧急气管切开术,术后患者呼吸困难好转,未完全缓解。鼻窦CT示:鼻骨形态欠自然,未见明显骨折线(图2)。胸部三维CT示:气管壁增厚、形态欠自然,右侧支气管中间段管腔明显狭窄(图3)。颈部CT示:喉术后:喉前皮下软组织结构紊乱,局部甲状软骨前缘骨质不连续,气管前方软组织变薄(邻近层面存在运动伪影,部分结构显示不清),双侧杓状软骨显示可,双侧声带形态欠规整,声门裂及声门下狭窄。支气管镜检查示:隆突变钝,各级支气管管腔狭窄,呼气相明显。经风湿免疫科、呼吸内科会诊,均考虑复发性多软骨炎。呼吸科会诊:考虑患者病情较重,已出现多发多级气管软骨塌陷,建议上级医院置入“Y”型气管支架治疗。与患者家属充分沟通病情及会诊意见,家属决定放弃支架植入,回当地治疗,患者于2018年1月23日出院,此后患者未再回我院復查,具体恢复情况不详。
2讨论
复发性多软骨炎(relapsing polychondritis,RP)是一种少见的由免疫介导的结缔组织疾病,呈缓解-复发型、进行性炎症临床表现,其表现为关节炎、眼疾、听力减退、消化系统疾病、皮肤损伤、心脏瓣膜功能不全、血管炎等,反复炎症将导致相应的组织退行性变。RP的病因尚不明确,研究表明[1,2],其与自身免疫反应介导的Ⅱ型胶原蛋白有关,而该蛋白在软骨和巩膜中含量丰富。复发性多软骨炎发病在中年人中多见,发病高峰年龄在40~55岁,女性较男性略微高发,也有儿童及老年人患病的报道,但无种族特异性。美国RP的年发病率为3.5/100万[3],英国RP发病率为1/140万,标准化死亡率为2.16[4]。目前国内尚无较大样本的流行病学研究。
复发性多软骨炎的临床表现有多样性和非特异性,该病的特征性病变为软骨炎,也是诊断RP的必要条件。软骨结构的反复炎症反应最终将导致局部退化和萎缩。研究表明[5],在疾病初期有一半的患者并无软骨炎的表现,因此该病极易误诊,延误诊断的时间为数月甚至数年不等。其中,最常见的临床表现是耳廓软骨炎,可有单耳或双耳耳廓软骨区域的红、肿、热、痛,而耳垂无异常,病程持续数天或更长,自发消退后可能间隔复发,最终导至软骨溶解,外耳畸形呈菜花样耳。病变耳廓质地也不尽相同,大部分表现为质软,也有少部分为质硬,后者是由结缔组织的钙化或骨化引起。有10%左右的患者会发生菜花耳畸形,而表现为感音神经性听力丧失和耳鸣的患者较少见[6]。鼻软骨炎比耳软骨炎少见,特点是鼻部软骨炎症引起疼痛及鼻塞的感觉,最终导致不可逆的鞍鼻畸形。RP在鼻部也可表现为鼻出血、鼻溢液和鼻痂。在疾病进程中多达一半的病人会出现呼吸道相关问题,而呼吸道并发症和下呼吸道感染是RP中最常见的死亡原因[7]。一半以上患者可发生喉软骨炎,表现为声音嘶哑、气管环压痛,咳嗽,呼吸困难和喘鸣。呼吸道症状通常是由气道炎症引起,软骨结构支架逐步破坏,最终导致气道的动态塌陷,尤其是在强制呼气时,表现更为明显,可通过肺功能测试和动态CT进行鉴别[8]。慢性喉气管和支气管软骨炎可导致危及生命的气道狭窄,有35%的RP患者发生肋软骨炎,但很少表现为首发症状,其表现为胸骨后疼痛,严重者影响呼吸[1,5]。
关节炎是RP第二常见表现。RP患者关节病变表现呈非对称性、间断性炎症,是一种非破坏性、非侵蚀性、血清阴性的少/多关节炎[9]。心血管系统并发症是继呼吸系统疾病的第二大常见死因,累及心血管系统最常见的表现是主动脉根部扩张引起的主动脉返流[1]。非特异性皮肤病表现,如紫癜,丘疹和结节,在RP病人中也很常见。RP患者的眼部表现最常见的是巩膜炎、结膜炎。角膜炎和葡萄膜炎的报道较少。较严重的眼部表现比较少见,包括视网膜动脉和静脉阻塞、视功能减退神经炎、视网膜病变和视网膜脱离[2,5]。疾病很少发生于神经系统,一旦发生,死亡率较高,最常见表现为第Ⅴ和第Ⅶ脑神经麻痹,也有表现为脑膜炎、脑炎、中风和动脉瘤[5]。有证据表明[10],RP是痴呆症的罕见病因。肾脏受累较罕见,预后较差,最常见的病理生理表现是系膜增生,其次是节段性坏死性肾小球肾炎。另有研究报道[11],肾脏受累存的病理生理表现为IgA肾病和肾小管间质性肾炎,可能伴随有炎症性肠病和自主神经功能障碍但其发生率仍不清楚。
目前复发性多软骨炎的诊断多依据1976年提出的McAdam's诊断标准,分别为主要标准,包括耳廓软骨发炎、鼻软骨发炎、喉气管软骨发炎,和次要诊断,包括葡萄膜炎、主膜炎、巩膜炎或葡萄膜炎、听力损失、听力损失、血清反应阴性的多发性关节炎,而诊断复发性多软骨炎需要两个主要标准或一个主要标准+两个次要标准。或者依据1979年经Damnian和Levine修订的标准,至少具备McAdam's诊断标准中的3项,无须病理证实;或至少具备McAdam's诊断标准中的1项,并有病理证实,或病变涉及两处各自独立的解剖部位,并对激素和(或)对氨苯砜治疗有效。在临床证据不足时,可考虑软骨活检,组织学显示软骨组织碎片被纤维结缔组织包围,单核炎性浸润,纤维化伴软骨周炎的反应多提示为复发性多软骨炎。对疑似RP患者的初步临床评估应该包括ESR和CRP,可用于评估疾病活动及对治疗的反应。复发性多软骨炎活动指数已经确认用来评估疾病的活动性并且可以用来量化疾病严重性和辅助治疗的决策[12]。现在RP的治疗主要是对症治疗,由于该病较罕见,没有大样本的随机对照研究,因此目前没有建立标准的治疗方案。患者的初始治疗由非甾体抗炎药(NSAIDs)组成/激素和或免疫抑制治疗。治疗可减轻RP的即刻症状,但长期治疗并不能阻止疾病的进展。新的治疗方法已经应用于临床,如TNF-ɑ阻滞剂用于治疗严重或难治性患者已取得不同程度的成功,此类生物制剂有可能减缓甚至终止RP的自然进程[13]。
随着人们对RP疾病认识的深入,RP患者的预后近年来得到改善。由5年生存率70%[7]到10年生存率91%[14]。有研究表明[4],患有明顯气道受累的RP患者预后差,其最常见的死亡原因是喉气管支气管疾病/感染或心血管并发症。而决定患者预后的关键是及时的诊断与治疗干预,该病由于临床表现多样,缺乏特异性,其中大量患者首诊于耳鼻喉科,而治疗主要在风湿免疫科或呼吸内科等科室,很多耳鼻喉科年轻医师对于该疾病认识不足,在疾病早期极易误诊及漏诊,延误治疗的最佳时期。本病例患者在疾病初期表现为声嘶、呼吸困难,电子喉镜表现为喉室饱满、左侧声带固定,颈部增强CT示:左侧喉旁间隙肿物,边界欠清,从临床症状、体征及影像学检查都极易误诊为肿瘤,最终术中及术后病理证实为非特异性炎症。此后患者反复出现咳嗽、呼吸困难、鼻塞、流涕等,均在当地诊为上呼吸道感染、气管炎,并未进一步检查及治疗。当第二次就诊我院时,患者已出现严重的喉软骨变形、气管软骨塌陷、鼻软骨塌陷等不可逆改变。因此,耳鼻喉科医师应从该病例中汲取经验与教训,在接诊有类似耳廓软骨炎、鼻软骨炎或喉气管软骨炎症状的患者时,应注意仔细询问相关病史,包括其他易累及器官,关节、眼、皮肤等,完善相关检查,及时请相关科室会诊,避免诊断思维局限而延误诊断与治疗。
参考文献:
[1]Kingdon J,Roscamp J,Sangle S,et al.Relapsing polychondritis:a clinical review for rheumatologists[J]. Rheumatology(Oxford),2018,57(9):1525-1532.
[2]Letko E,Zafirakis P,Baltatzis S,et al.Relapsing polychondritis:A clinical review[J].Seminars in Arthritis&Rheumatism,2002,31(6):384-395.
[3] Alatas F,Ozkan R,Metintas M,et al.Relapsing polychondritis.Respirology,2003,8(1):99-103. [4]Hazra N,Dregan A,Charlton J,et al.Incidence and mortality of relapsing polychondritis in the UK:a population-based cohort study[J].Rheumatology(Oxford),2015,54(12):2181-2187.
[5]Puéchal X,Terrier B,Mouthon L,et al.Relapsing polychondritis[J].Joint Bone Spine,2014,81(2):118-124.
[6]Smylie A,Malhotra N,Brassard A.Relapsing Polychondritis:A Review and Guide for the Dermatologist[J].American Journal of Clinical Dermatology,2016,18(1):1-10.
[7]Rafeq S,Trentham D,Ernst A.Pulmonary Manifestations of Relapsing Polychondritis[J].Clinics in Chest Medicine,2010,31(3):513-518.
[8]Church RE.Relapsing polychondritis[J].Laryngoscope,1972,82(5):891-898.
[9]Lahmer T,Treiber M,Werder AV,et al.Relapsing polychondritis:An autoimmune disease with many faces[J].Autoimmunity Reviews,2010,9(8):540-546.
[10]Hong JC.Relapsing Polychondritis with Central Nervous System Involvement:Experience of Three Different Cases in a Single Center[J].Journal of Korean Medical Science,2016,31(11):1846.
[11]Aguilar MC,Lonngi M,de-la-Torre A.Tubulointerstitial Nephritis and Uveitis Syndrome:Case Report and Review of the Literature[J].Ocul Immunol Inflamm,2016,24(4):415-421.
[12]Arnaud L,Devilliers H,Peng SL,et al.The Relapsing Polychondritis Disease Activity Index:Development of a disease activity score for relapsing polychondritis[J].Autoimmunity Reviews,2012,12(2):204-209.
[13]Lekpa FK,Kraus VB,Chevalier X.Biologics in Relapsing Polychondritis:A Literature Review[J].Seminars in Arthritis and Rheumatism,2012,41(5):712-719.
[14]Dion J,Costedoatchalumeau N,Sène D,et al.Relapsing polychondritis can be characterized by 3 different clinical phenotypes:Analysis of a recent series of 142 patients[J].Arthritis&Rheumatology,2016,68(12):2992-3001.
收稿日期:2019-9-16;修回日期:2019-10-12
編辑/杜帆
