基于SNP遗传图谱定位甘蓝型油菜分枝角度QTL
2019-12-25汪文祥梅德圣成洪涛朱琳琳
汪文祥 储 文 梅德圣 成洪涛 朱琳琳 付 丽 胡 琼 刘 佳,*
基于SNP遗传图谱定位甘蓝型油菜分枝角度QTL
汪文祥1储 文1梅德圣1成洪涛1朱琳琳2付 丽1胡 琼1刘 佳1,*
1中国农业科学院油料作物研究所/ 农业农村部油料作物生物学与遗传育种重点实验室, 湖北武汉 430062;2南漳县植保站, 湖北襄阳 441500
分枝角度是油菜株型的重要性状, 与油菜的耐密植性密切相关。本研究利用油菜分枝角差异显著的育种亲本材料1098B (分枝角小)和R2 (分枝角大)杂交获得F1, 通过小孢子培养获得含163份株系的DH群体。以油菜60K SNP芯片进行DH群体基因分型, 构建高密度遗传图谱, 并利用QTL Cartographer 2.5对2个环境下油菜顶端分枝角和基部分枝角进行QTL分析。结果表明, 构建的高密度遗传图谱覆盖甘蓝型油菜19条染色体, 包含9521个多态性SNP标记, 1442个簇(bin), 覆盖基因组长度为2544.07 cM, 相邻簇(bin)之间平均距离为1.76 cM。在此图谱基础上采用复合区间作图法(CIM), 在2个环境下检测到17个分枝角度QTL, 分别位于A01、A02、A03、A06、A09、C02、C03、C04、C06和C08染色体上, 单个QTL解释的表型变异为6.39%~21.78%。用比较基因组方法与拟南芥分枝角度同源基因区间比对, 鉴定出其中6个QTL的12个候选基因。其中位于A03连锁群QTL在2年的试验中被重复检测到, 根据物理位置和基因组信息推测为分枝角度的候选基因。这些QTL和候选基因将为油菜分枝角度的遗传改良提供有用的信息。
甘蓝型油菜; 分枝角度; 60K SNP芯片; QTL位点; 候选基因
油菜是我国重要的油料作物, 每年提供450万吨左右食用油, 占全国植物油总产的40%以上[1]。现阶段, 油菜单产水平低和机械化生产普及率不高是限制我国油菜产业发展的两大瓶颈。为增加单产、提高油菜机械化生产效率, 改良现有品种的株型、提高油菜的种植密度是一条有效途径。改良油菜株型不仅可以促进光能和肥料利用率, 提高品种高密度耐性, 降低病害, 增加群体产量[2], 还有利于增强抗倒性和机械化生产, 通过资源高效化利用实现低耗高产[3]。
随着甘蓝型油菜全基因组测序的完成[4-5]、高通量SNP芯片技术的完善和分析成本的下降, 油菜60K SNP芯片分型分析成为油菜遗传研究的重要工具[6-11]。近几年, 利用油菜60K SNP芯片对油菜产量及品质等性状的遗传定位做了大量的研究[12-14]。Liu等[6]利用油菜芯片对RIL群体进行分型, 对种子颜色、木质素、纤维素和半纤维素等性状进行QTL定位。Sun等[15]利用520份油菜资源群体对株高性状进行全基因组关联分析, 鉴定到68个显著的SNP位点, 超过70%的位点与已发表的9个遗传群体中重合。Fu等[16]利用DH群体和永久F2群体对角果长和千粒重性状进行联合QTL分析, 将A9染色体的角果长和千粒重区间缩小到1.14 Mb内。油菜SNP芯片技术的利用, 提高了遗传图谱构建精密度, 为油菜数量性状的研究提供更方便、快捷的途径。
油菜分枝角度是构建理想株型的重要因素, 紧凑型株型有利于密植, 有利于提高群体的产量和抗倒伏性, 减少病害[17-24]。Liu等[18]最早利用143份自然群体对分枝角度进行全基因组关联分析, 获得6个显著关联的基因组区域, 鉴定出其中4个区域内候选基因。Sun等[19]利用520份自然群体进行全基因组关联分析, 共获得52个分枝角度相关位点, 总共可解释表型变异的51.1%, 并对52个位点进行了候选基因分析。Wang等[20]利用分枝角差异显著亲本构建F2群体, 鉴定F2代所有单株的分枝角表型, 挑选极端单株进行混池, 利用BSA-seq方法确定A6染色体定位区间内生长素合成相关基因为候选基因。Shen等[22]利用包含208个系的DH群体定位了17个油菜分枝角度QTL, 3个主效的QTL各能解释约10%表型变异, 并在QTL区间获得27个候选基因。上述研究表明, 油菜分枝角度受多基因控制, 油菜分枝角度性状的遗传解析对油菜品种的遗传改良具有重要意义。
本研究利用油菜60K SNP芯片构建高密度遗传连锁图谱, 对不同部位油菜分枝角度性状进行QTL定位分析。收集整理前人研究油菜分枝角度基因信息, 并利用甘蓝型油菜基因组序列, 根据QTL区间物理位置及拟南芥功能基因组信息筛选可能的候选基因, 为明确关键QTL和克隆候选基因奠定基础。
1 材料与方法
1.1 材料
以分枝角度小的甘蓝型油菜品系1019B和分枝角度大的R2为亲本, 手工配置F1, 通过小孢子培养获得DH群体。选取其中163个DH系进行SNP标记分型, 用以构建高密度SNP遗传连锁图谱。所有材料均由中国农业科学院油料作物研究所提供及保存。
1.2 田间试验
2013年9月与2014年9月, 将亲本及DH系群体种植于中国农业科学院油料作物研究所阳逻试验基地, 分别记录为WH2014和WH2015。随机区组设计2个重复, 每个小区3行, 每行18株, 行距0.33 m,株距0.10 m。田间管理同常规生产, 确保所有样本的外部生长环境一致, 待成熟后统计数据。
1.3 性状考察
油菜成熟后, 从每个株系选取正常生长的5株, 剪取连有油菜上部第一分枝(顶枝)和基部第一分枝(基枝)的茎段, 通过数字图像采集法[17]获取顶端分枝角和基部分枝角图像文件, 将其导入AutoCAD软件, 利用角度工具标注角度, 记录到Microsoft Excel文档中。
1.4 遗传连锁图谱及QTL分析
利用油菜60K Illumina InfiniumSNP芯片对亲本(1019B和R2)及DH群体进行SNP基因分型。采用GenomeStudio (Illumina公司)软件分析SNP基因型, 排除低于0.05的最小基因型频率(minor allele frequency, MAF), SNP得率(call frequency)小于80%, 筛选在亲本间具有多态性的标记, 利用SNP芯片序列信息和法国公布的甘蓝型油菜品种“”的基因组序列信息进行BlastN比对, E-value阈值为1×10–15, 获得唯一位置的SNP标记信息, 最终获得9521个高质量SNP标记用于后续分析。
利用MSTmap软件[25]和JoinMap 4.0软件[26]构建遗传图谱, 通过两两标记之间最小重组频率计算每个连锁群上标记顺序, 构建高密度bin-map遗传图谱。利用Windows QTL Cartographer 2.5软件[27]中复合区间作图(Composite Interval Mapping, CIM)法对2个环境下基部分枝角和顶端分枝角进行单环境QTL检测。运行CIM时, 选用1 cM的步长(walking speed), 同时设置5个标记作为余因子, 采用模型6, 对每个性状分别进行1000次排列测验(permutation test), 显著水平0.05来判断是否存在QTL。运行结果同时给出性状QTL的加性效应和表型贡献率。以q加顶端分枝角度英文缩写“顶端分”或基部分枝角度英文缩写“基部分”, 再加上染色体编号及QTL序号命名QTL。在染色体相同的位置重复的QTL, 且加性效应方向一致, 认为是同一QTL。采用MapChart 2.3[28]绘制QTL定位遗传连锁图。
1.5 候选基因筛选
为了筛选出分枝角度相关的候选基因, 基于油菜基因组和拟南芥基因组序列进化的同源性[29], 在甘蓝型油菜基因组[4]上查询检测到的QTL置信区间对应的序列, 然后与Liu等[15]、Sun等[25]、Li等[21]和Shen等[22]搜索出的拟南芥分枝角度相关基因进行BlastN比对, 将E值设定为1×10–20, 最后筛选出每个QTL置信区间内匹配E值小于阈值的候选基因。
2 结果与分析
2.1 双亲及DH群体分枝角度表型变异
在两年的环境中, 2个亲本的顶端分枝角和基部分枝角差异均极显著(表1)。1019B顶端分枝角和基部分枝角均较小, R2顶端分枝角和基部分枝角均较大。DH群体顶端分枝角和基部分枝角均呈连续性分布, 其中一些株系表现出明显的超亲分离现象, 平均变异幅度分别为22.98°~63.99°和20.76°~50.22° (图1),平均值介于两亲本之间; 顶端分枝角和基部分枝角的变异系数都大于10, 说明顶端分枝角和基部分枝角都具有较大的改良潜力。
相关性分析表明, 在2014年和2015年的2个环境中顶端分枝角和基部分枝角表现均呈极显著正相关(表2), 相关系数分别为0.362和0.411, 说明油菜分枝角性状遗传稳定, 但受一定环境影响。在2个生长周期里, 顶端分枝角和基部分枝角呈极显著正相关, 相关系数分别为0.542和0.586, 说明油菜顶端分枝角和基部分枝角相关性较强, 具有一致性。
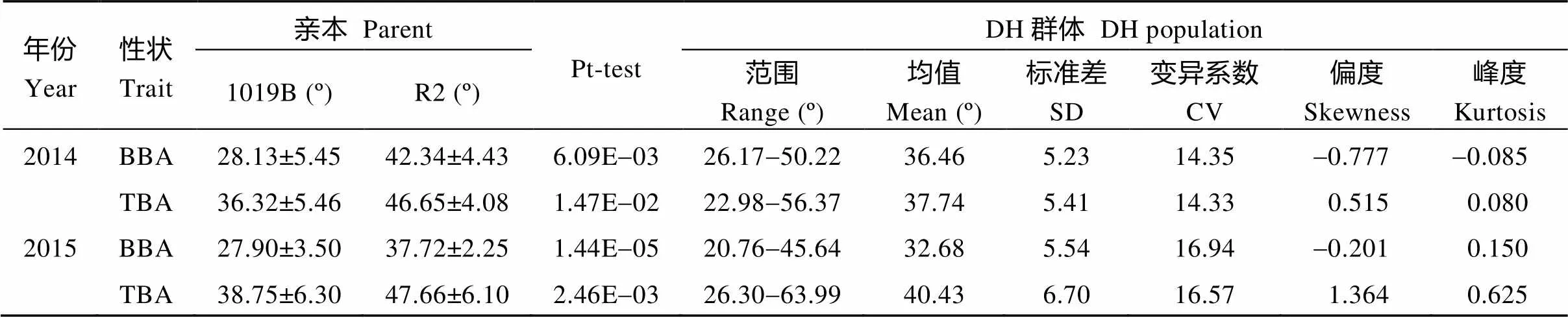
表1 亲本及DH群体分枝角度性状两年的表型分析
BBA: 基部分枝角; TBA: 顶端分枝角。BBA: basal branch angle; TBA: top branch angle.
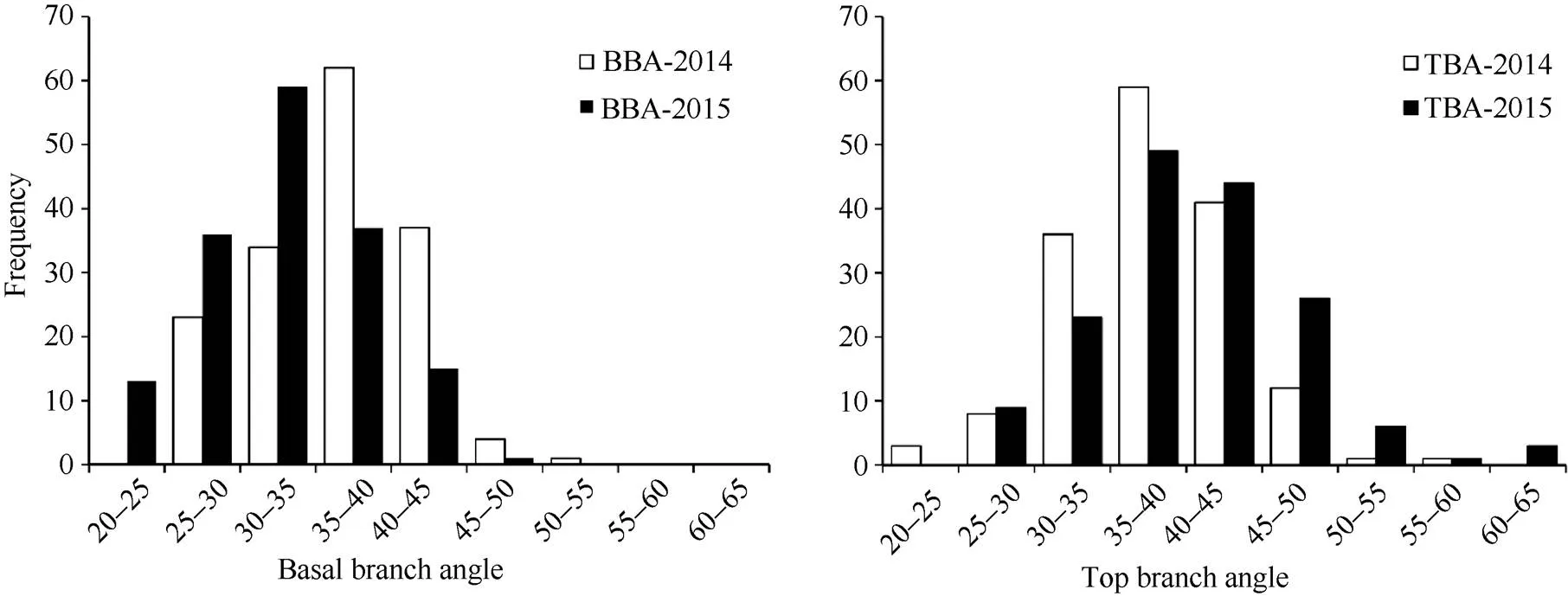
图1 油菜1019B×R2 DH群体分枝角度性状(BBA和TBA)两年的频率分布图

表2 在2014年和2015年甘蓝型油菜DH群体分枝角度性状的相关系数
BBA: 基部分枝角; TBA: 顶端分枝角。**代表在0.01显著水平。
BBA: basal branch angle; TBA: top branch angle.**denote significant correlation at the 0.01 probability level.
2.2 高密度遗传图构建
利用油菜SNP芯片, 检测DH群体的基因型, 共得到9521个高质量多态性SNP标记。基于这些SNP标记, 利用MSTmap软件构建DH群体的重组区块图谱共得到1442个Bin, 并利用这些Bin构建的遗传连锁图谱总长度为2544.07 cM, 标记间的平均距离1.76 cM, 标记间最大距离为30.59 cM, 最小距离0.06 cM; 其中A10染色体上标记间的平均遗传距离最小(1.03 cM), C09染色体上标记间平均遗传距离最大(3.73 cM)(图2)。

图2 SNP标记簇在连锁群上分布图
纵坐标显示甘蓝型油菜基因组19个连锁群中的每一个SNP簇的遗传距离。
The ordinate shows the genetic distance along each of the 19 linkage groups corresponding togenome.
2.3 油菜分枝角度QTL分析
利用软件Windows QTL Cartographer 2.5对顶端分枝角和基部分枝角性状进行QTL分析, 共检测到17个QTL, 分布于A01、A02、A03、A06、A09、C02、C03、C04、C06和C08染色体上, 单个QTL可解释的表型变异为6.39%~21.78% (表3和图3)。其中检测到6个基部分枝角QTL, 单个QTL解释的表型变异为8.07%~15.10%; 检测到11个顶端分枝角QTL, 单个QTL解释的表型变异为6.39%~21.78%。在2年内重复检测到基部分枝角性状显著效应的QTL和, 与在一个环境中检测到的顶端分枝角性状QTL ()在同一个置信区间, 在3个环境中解释的表型变异为8.28%~11.79%, 加性效应值为1.74~2.03。另外, 在同一环境中检测到顶端分枝角和基部分枝角共同的QTL (和), 解释表型变异8.78%~8.85%。

表3 2个环境中检测到的顶端分枝角和基部分枝角QTL
BBA: 基部分枝角; TBA: 顶端分枝角。BBA: basal branch angle; TBA: top branch angle.
2.4 分枝角度候选基因筛选
将17个QTL置信区间序列与前人报道的181个分枝角度相关基因分别比对, 在其中6个QTL区间内检测到12个候选基因。在两年均检测到的QTL (、和)置信区间内发现生长素转运相关的基因, 其物理位置离整合后的QTL峰值非常近, 该基因也存在于Sun等[19]定位的分枝角度QTL区间内。在基部分枝角度QTL (置信区间内筛选到生长素相关的基因; 在顶端分枝角度2个QTL (和置信区间内分别筛选出2个分枝角度候选基因。另外, 在顶端分枝角度QTL (置信区间内筛选到4个生长素相关候选基因; 与控制水稻分蘖角基因同源的基因也在顶端分枝角的QTL ()区间内检测到(表4)。
3 讨论
株型紧凑和抗倒伏是实现油菜全程机械化栽培的重要性状。本研究通过高通量SNP标记构建高密度遗传图谱, 鉴定获得了油菜分枝角的QTL及两侧与性状连锁的标记, 为油菜分枝角度的分子标记辅助育种提供了依据。我们前期研究发现, 油菜从基部到顶部的分枝角度并不完全一样, 有逐渐增大的趋势[30]。不同材料间从下到上分枝角度变化具有同样的趋势, 这是由油菜多分枝生长发育习性决定的。本研究同时调查顶部和基部的分枝角度, 可以对这一性状进行更全面的分析。本研究获得的油菜分枝角度QTL多数与已研究报道的QTL[18-22]相吻合, 还鉴定到一些新的QTL; 2个环境重复检测到位于A03连锁群的QTL效应稳定, 与前人利用关联分析方法获得的peak-SNP ()位点相同。顶端分枝角度和基部分枝角在同一环境中均在C08连锁群定位到重合的QTL, 并且表型数据表明顶端分枝角和基部分枝角具有显著相关性, 说明可能存在同时控制油菜上、下分枝角度的基因。

图3 甘蓝型油菜顶端分枝角和基部分枝角QTL在连锁群上分布图

表4 在甘蓝型油菜分枝角QTL置信区间比对获得的候选基因
随着高通量测序技术的发展, 油菜参考基因组信息的公布[4-5], 油菜基因组上大量的SSR、Indel、SNP等标记被发掘出来[31-32]。SNP标记因为数量多、分布广, 在基因组中密度更高和分布更均匀, 已实现SNP基因型分型的高通量、快速和自动化检测, 使不同研究者间的数据整合分析成为现实[33]。本研究构建的高密度遗传图谱, 标记间平均距离1.76 cM, 是较精密的甘蓝型油菜遗传图谱之一。利用该图谱获得的分枝角性状相关SNP标记, 有助于进一步发展加密QTL区间的分子标记, 用于进一步精细定位和功能基因克隆。
利用油菜基因组序列信息及SNP标记的侧翼序列, 从分枝角度的QTL置信区段内, 借助和拟南芥基因组间同源区间序列比对, 筛选出12个油菜分枝角候选基因。其中坐落于A03染色体上的候选基因, 与拟南芥的基因同源。据拟南芥基因的功能注释,基因正向调节生长素, 在生长素的极性运输中通过正确的蛋白分泌与定位来实现其功能, 在植物分枝数或分生组织的建成中发挥着重要作用[34]。位于C04染色体的候选基因, 该基因的拟南芥同源基因是。是控制水稻分蘖角度增加的主效基因, 具有显性效应[35]; 在玉米[36]、小麦和桃树[37]中也已分别克隆和验证了基因的功能。因此,类基因在植物中具有广泛的调节侧生分枝水平生长的功能[34]。在定位到的分枝角度QTL区间内还鉴定到生长激素相关的基因、、和。此外, 本研究还在QTL区间内发现许多未知功能的候选基因, 可能存在油菜特有的分枝角度的功能基因, 有待进一步挖掘。
4 结论
本研究构建了包含19条染色体的油菜高密度遗传图谱, 该图谱含9521个多态性标记, 经过整合为1442个簇, 相邻簇之间平均距离为1.76 cM。定位到位于A01、A02、A03、A06、A09、C02、C03、C04、C06和C08染色体上的17个分枝角度QTL, 其中位于A03染色体上的QTL能在两年环境下被重复检测到, 贡献率为9.31%~11.80%, 在A03染色体上重复检测到的QTL 置信区间获得1个与分枝角度相关的候选基因, 在C04染色体上的QTL置信区间获得1个控制水稻分蘖角度的候选基因; 分别在染色体A01、A09、C03和C06上QTL中检测到生长激素相关基因、、和。本研究为进一步鉴定和克隆油菜分枝角度功能基因奠定了基础。
[1] 王汉中, 殷艳. 我国油料产业形势分析与发展对策建议. 中国油料作物学报, 2014, 36: 414–421. Wang H Z, Yin Y. Analysis and strategy for oil crop industry in China., 2014, 36: 414–421 (in Chinese with English abstract).
[2] Cai G Q, Yang Q Y, Chen H, Yang Q, Zhang C Y, Fan C C, Zhou Y M. Genetic dissection of plant architecture and yield-related traits in., 2016, 6: 21625.
[3] Wang Y H, Li J Y. Molecular basis of plant architecture., 2008, 59: 253–279.
[4] Chalhoub B, Denoeud F, Liu S Y, Parkin I A, Tang H B, Wang X Y, Chiquet J, Belcram H, Tong C B, Samans B, Corréa M, Da Silva C, Just J, Falentin C, Koh C S, Le Clainche I, Bernard M, Bento P, Noel B, Labadie K, Alberti A, Charles M, Arnaud D, Guo H, Daviaud C, Alamery S, Jabbari K, Zhao M X, Edger P P, Chelaifa H, Tack D, Lassalle G, Mestiri I, Schnel N, Le Paslier M C, Fan G, Renault V, Bayer P E, Golicz A A, Manoli S, Lee T H, Thi V H, Chalabi S, Hu Q, Fan C, Tollenaere R, Lu Y, Battail C, Shen J, Sidebottom C H, Wang X, Canaguier A, Chauveau A, Bérard A, Deniot G, Guan M, Liu Z, Sun F, Lim Y P, Lyons E, Town C D, Bancroft I, Wang X, Meng J, Ma J, Pires J C, King G J, Brunel D, Delourme R, Renard M, Aury J M, Adams K L, Batley J, Snowdon R J, Tost J, Edwards D, Zhou Y, Hua W, Sharpe A G, Paterson A H, Guan C, Wincker P. Early allopolyploid evolution in the post-Neolithicoilseed genome., 2014, 345: 950–953.
[5] Sun F M, Fan G Y, Hu Q, Zhou Y M, Guan M, Tong C B, Li J N, Du D Z, Qi C K, Jiang L C, Liu W Q, Huang S M, Chen W B, Yu J Y, Mei D S, Meng J L, Zeng P, Shi J Q, Liu K D, Wang X, Wang X F, Long Y, Liang X M, Hu Z Y, Huang G D, Dong C H, Zhang H, Li J, Zhang Y L, Li L W, Shi C C, Wang J H, Lee M S, Guan C Y, Xu X, Liu S Y, Liu X, Chalhoub B, Hua W, Wang H Z. The high-quality genome ofcultivar ‘ZS11’ reveals the introgression history in semi-winter morphotype., 2017, 92: 452–468.
[6] Liu L Z, Qu C M, Wittkop B, Yi B, Xiao Y, He Y J, Snowdon R J, Li J N. A high-density SNP map for accurate mapping of seed fibre QTL inL., 2013, 8: e83052.
[7] Wang N, Li F, Chen B Y, Xu K, Yan G X, Qiao J W, Li J, Gao G Z, Bancroft L, Meng J L, King G, Wu X M. Genome-wide investigation of genetic changes during modern breeding of., 2014, 127: 1817–1829.
[8] 张凤启, 刘越英, 程晓辉, 童超波, 董彩华, 唐敏强, 黄军艳, 刘胜毅. 利用高密度 SNP 标记定位甘蓝型油菜株高 QTL. 中国油料作物学报, 2014, 36: 695–700. Zhang Q F, Liu Y Y, Cheng X H, Tong C B, Dong C H, Tang M Q, Huang J Y, Liu S Y. QTL mapping of plant height using high density SNP markers in., 2014, 36: 695–700 (in Chinese with English abstract).
[9] Xu L P, Hu K N, Zhang Z Q, Guan C Y, Shen S, Hua W, Li J N, Wen J, Yi B, Shen J X, Ma C Z, Tu J X, Fu T D. Genome-wide association study reveals the genetic architecture of flowering time in rapeseed (L.)., 2015, 23: 43–52.
[10] Li F, Chen B Y, Xu K, Gao G Z, Yan G X, Qiao J W, Li J, Li H, Li L X, Xiao X, Zhang T Y, Nishio T, Wu X M. A genome-wide association study of plant height and primary branch number in rapeseed ()., 2016, 242: 169–177.
[11] Liu J, Wang J, Wang H, Wang W X, Zhou R J, Mei D S, Chen H T, Yang J, Raman H, Hu Q. Multigenic control of pod shattering resistance in Chinese rapeseed germplasm revealed by genome-wide association and linkage analyses., 2016, 7: 1058.
[12] Luo X, Ma C Z, Yue Y, Hu K N, Li Y Y, Duan Z Q, Wu M, Tu J X, Shen J X, Yi B, Fu T D. Unravelling the complex trait of harvest index in rapeseed (L.) with association mapping., 2015, 16: 379.
[13] Lu K, Peng L, Zhang C, Lu J H, Yang B, Xiao Z C, Liang Y, Xu X F, Qu C M, Zhang K, Liu L Z, Zhu Q L, Fu M L, Yuan X Y, Li J N. Genome-wide association and transcriptome analyses reveal candidate genes underlying yield-determining traits in., 2017, 8: 206.
[14] Liu S, Fan C C, Li J N, Cai G Q, Yang Q Y, Wu J, Yi X Q, Zhang C Y, Zhou Y M. A genome-wide association study reveals novel elite allelic variations in seed oil content of., 2016, 129: 1203–1215.
[15] Sun C M, Wang B Q, Yan L, Hu K N, Liu S, Zhou Y M, Guan C Y, Zhang Z Q, Li J N, Chen S, Wen J, Ma C Z, Tu J X, Shen J X, Fu T D, Yi B. Genome-wide association study provides insight into the genetic control of plant height in rapeseed (L.)., 2016, 7: 1102.
[16] Fu Y, Wei D Y, Dong H L, He Y J, Cui Y X, Mei J Q, Wan H F, Li J N, Snowdon R, Friedt W, Li R X, Qian W. Comparative quantitative trait loci for silique length and seed weight in., 2015, 5: 14407.
[17] 汪文祥, 胡琼, 梅德圣, 李云昌, 王会, 王军, 付丽, 刘佳. 基于图像处理的油菜分枝及角果着生角度测量方法. 中国油料作物学报, 2015, 37: 566–570. Wang W X, Hu Q, Mei D S, Li Y C, Wang H, Wang J, Fu L, Liu J. Evaluation of branch and pod angle measurement based on digital images fromL., 2015, 37: 566–570 (in Chinese with English abstract).
[18] Liu J, Wang W, Mei D, Wang H, Fu L, Liu D, Li Y, Hu Q. Characterising variation of branch angle and genome-wide association mapping in rapeseed (L.)., 2016, 7: 21.
[19] Sun C, Wang B, Wang X. Genome-wide association study dissecting the genetic architecture underlying the branch angle trait in rapeseed (L.)., 2016, 6: 33673.
[20] Wang H, Cheng H T, Wang W X, Liu J, Hao M Y, Mei D S, Zhou R J, Fu L, Hu Q. Identification ofas a candidate gene for branch angle inby QTL-seq., 2016, 6: 38493.
[21] Li H G, Zhang L P, Hu J H, Zhang F G, Chen B Y, Xu K, Gao G Z, Li H, Zhang T Y, Li Z Y, Wu X M. Genome-wide association mapping reveals the genetic control underlying branch angle in rapeseed (L.)., 2017, 8: 1054.
[22] Shen Y S, Yang Y, Xu E S, Ge X H, Xiang Y, Li Z Y. Novel and major QTL for branch angle detected by using DH population from an exotic introgression in rapeseed (L.)., 2018, 131: 67–78.
[23] 张倩. 甘蓝型油菜主要株型性状的遗传分析和QTL初步定位. 西南大学硕士学位论文, 重庆, 2013. Zhang Q. Genetic Effects Analysis and QTL Mapping of Major Plant-type Traits inL. MS Thesis of Southwest University, Chongqing, China, 2013 (in Chinese with English abstract).
[24] 汪文祥, 胡琼, 梅德圣, 李云昌, 周日金, 王会, 成洪涛, 付丽, 刘佳. 甘蓝型油菜分枝角度主基因+多基因混合遗传模型及遗传效应. 作物学报, 2016, 42: 1103–1111. Wang W X, Hu Q, Mei D S, Li Y C, Zhou R J, Wang H, Cheng H T, Fu L, Liu J. Genetic effects of branch angle using mixture model of major gene plus polygeneL., 2016, 42: 1103–1111 (in Chinese with English abstract).
[25] Wu Y H, Bhat P R, Close T J, Lonardi S. Efficient and accurate construction of genetic linkage maps from the minimum spanning tree of a graph., 2008, 4: e1000212.
[26] Van Ooijen J W. JoinMap version 4.0: software for the calculation of genetic linkage maps in experimental populations. Netherlands: Wageningen University, 2006. https://www.kyazma.nl/ index.php/JoinMap/.
[27] Wang S C, Basten C J, Zeng Z B. Windows QTL Cartographer 2.5. Raleigh, NC, USA: Department of Statistics, North Carolina State University, 2012. http://statgen.ncsu.edu/qtlcart/WQTLCart. htm.
[28] Voorrips R E. MapChart: software for the graphical presentation of linkage maps and QTLs., 2002, 93: 77–78.
[29] Parkin I A P, Gulden S M, Sharpe A G, Lukens L, Trick M, Osborn T C, Lydiate D J. Segmental structure of thegenome based on comparative analysis with.. 2005, 171: 765–781.
[30] Wang W X, Hu Q, Mei D S, Wang J, Cheng H T, Wang H, Fu L, Liu J. Identification of compact germplasm resources suitable for high density cultivation inL., 2018, 3: 33–41.
[31] Li H T, Younas M, Wang X F, Li X M, Chen L, Zhao B, Chen X, Xu J S, Hou F, Hong B H, Liu G, Zhao H Y, Wu X L, Du H Z, Wu J S, Liu K D. Development of a core set of single-locus SSR markers for allotetraploid rapeseed (L.)., 2013, 126: 937–947.
[32] Shi J Q, Huang S M, Zhan J P, Yu J Y, Wang X F, Hua W, Liu S Y, Liu G H, Wang H Z. Genome-wide microsatellite characterization and marker development in the sequencedcrop species., 2014, 21: 53–68.
[33] Mason A S, Higgins E E, Snowdon R J, Batley J, Stein A, Werner C, Parkin I A. A user guide to the60K Illumina Infinium™ SNP genotyping array., 2017, 130: 621–633.
[34] Dehiwala-Liyanage C K. Functional Analysis ofGene in Arabidopsis. PhD Dissertation of Durham University, Durham, UK, 2011.
[35] Yu B S, Lin Z W, Li H X, Li X J, Li J Y, Wang Y H, Zhang X, Zhu Z F, Zhai W X, Wang X K, Xie D X, Sun C Q., a major quantitative trait locus controlling tiller angle in rice., 2007, 52: 891–898.
[36] Ku L X, Wei X M, Zhang S F, Zhang J, Guo S L, Chen Y H. Cloning and characterization of a putativeortholog associated with leaf angle in maize (L.)., 2011, 6: e20621.
[37] Dardick C, Callahan A, Horn R, Ruiz K, Zhebentyayeva T, Hollender C, Whitaker M, Abbott A, Scorza R. PpeTAC1 promotes the horizontal growth of branches in peach trees and is a member of a functionally conserved gene family found in diverse plants species., 2013, 75: 618–630.
Quantitative trait loci mapping for branch angle and candidate gene screening inL.
WANG Wen-Xiang1, CHU Wen1, MEI De-Sheng1, CHENG Hong-Tao1, ZHU Lin-Lin2, FU Li1, HU Qiong1, and LIU Jia1,*
1Oil Crops Research Institute, Chinese Academy of Agricultural Sciences / Key Laboratory of Biology and Genetic Improvement of Oil Crops of the Ministry of Agriculture and Rural Affairs, Wuhan 430062, Hubei, China;2Plant Protection Station of Nanzhang, Xiangyang 441500, Hubei, China
Branch angle is an important agronomic trait of plant architecture. In this study, 163 lines of a DH population derived from a cross between 1019B (compact type) and R2 (loose type) were genotyped by using 60K SNP array and a high-density genetic linkage map was constructed with 1442 bins inclusive of 9521 SNP markers to detect quantitative trait loic (QTL) for basal branch angle and top branch angle. The genetic map contained 19 lingkage groups with a total length of 2544.07 cM and an average distance between adjacent bin-markers of 1.76 cM. Totally, 17 QTL for branch angle were detected on chromosomes A01, A02, A03, A06, A09, C02, C03, C04, C06, and C08, respectively. The phenotypic variation accounted by a single locus was from 6.36% to 21.78%. Twelve candidate genes of branch angle were found underlying six QTL by comparing with homologous genesin. Candidate genewas close to the peak position of A03 QTL confidence interval, which was identified on chromosome A03 in both environments. These QTL and candidate genes provide useful information for the genetic modification of rapeseed branch angle.
oilseed rape; branch angle; 60K SNP array;QTL mapping; candidate gene
): 2018-03-22;
2018-08-20;
2018-09-09.
10.3724/SP.J.1006.2019.84042
通信作者(Corresponding author):刘佳, E-mail: liujia02@caas.cn, Tel: 027-86711556
E-mail: wangwenxiang@caas.cn, Tel: 027-86711556
本研究由中国农业科学院科技创新工程(Group No. 118), 国家农业现代产业技术体系建设专项(CARS-12), 湖北省科技创新工程和国家自然科学基金项目(31471535, 31771842)资助。
This study was supported by the Science and Technology Innovation Project of Chinese Academy of Agricultural Sciences (Group No. 118), the China Agriculture Research System (CARS-12), the Hubei Agricultural Science and Technology Innovation Center, and the National Natural Science Foundation of China (31471535, 31771842).
URL:http://kns.cnki.net/kcms/detail/11.1809.S.20180918.1125.004.html
