无菌医疗器械生产企业灭菌控制常见缺陷分析及监管要点梳理
2019-12-19唐剑通讯作者姚鹏
文/唐剑 通讯作者/姚鹏
无菌医疗器械是一种无活微生物的产品。当医疗器械必须以无菌的形式提供时,在其灭菌前应将各种非预期的微生物污染降至最低。灭菌的目的就是灭活微生物,将非无菌产品转变为无菌产品。灭菌过程作为无菌医疗器械生产的特殊过程,是医疗器械生产过程中需要定期验证和重点控制的过程[1]。环氧乙烷灭菌是医疗器械领域最常用的灭菌方法之一,目前大多数无菌医疗器械生产企业普遍采用环氧乙烷灭菌。本人自担任医疗器械GMP检查员工作以来,参与过多次无菌医疗器械GMP现场检查,现将本人近年来参与的现场检查中与环氧乙烷灭菌控制方面的监管经验分析总结如下。
1.环氧乙烷灭菌方法简析
环氧乙烷(EO)是一种烷化剂,穿透力强,可穿透绝大多数包装材料和聚合材料。广泛认同的成分包括纯EO和EO与二氧化碳或氮气的混合物。可应用于大多数不耐高温、不耐湿的包装材料和生物医用高分子材料的灭菌,被认为是一种灭菌效果最好的化学灭菌剂,可杀死所有微生物包括细菌以及芽孢[2]。但是,EO属于有毒、易燃、易爆物质,因此,在使用时要格外的小心。以下对EO灭菌的优缺点做一简要梳理,参见表1。
2.无菌医疗器械生产企业灭菌控制常见缺陷分析及监管要点梳理
2.1 灭菌控制常见不符合项特征及其判定依据
通过对2017~2018年来笔者实际参与的及从各地药监部门公开的共计40余家无菌医疗器械生产企业GMP检查结果进行汇总,调取其中与EO灭菌控制有关的不符合项,依据国家药监局规范性文件及相关法规标准,对各个不符合项按照特征类型及判定依据进行归类合并,最终形成灭菌确认、仓储管理、灭菌过程控制、灭菌残留控制、文件管理、设备管理、人员管理、销售管理、采购管理、留样管理10大方面的共性缺陷表(表2),从中可以看出,EO灭菌控制的常见共性缺陷数量众多,分布广泛,涵盖了采购、生产、检验、仓储、销售的生产质量管理体系全流程。反映出生产企业相关从业人员对于近年来国家药监局发布的配套指导原则、指南及相关行业标准具体要求的正确理解上还存在较大偏差,在执行上仍有很大改进空间(表2)。
2.2 共性缺陷类型占比分析

表1 EO灭菌的优缺点

表2 EO灭菌控制共性缺陷表
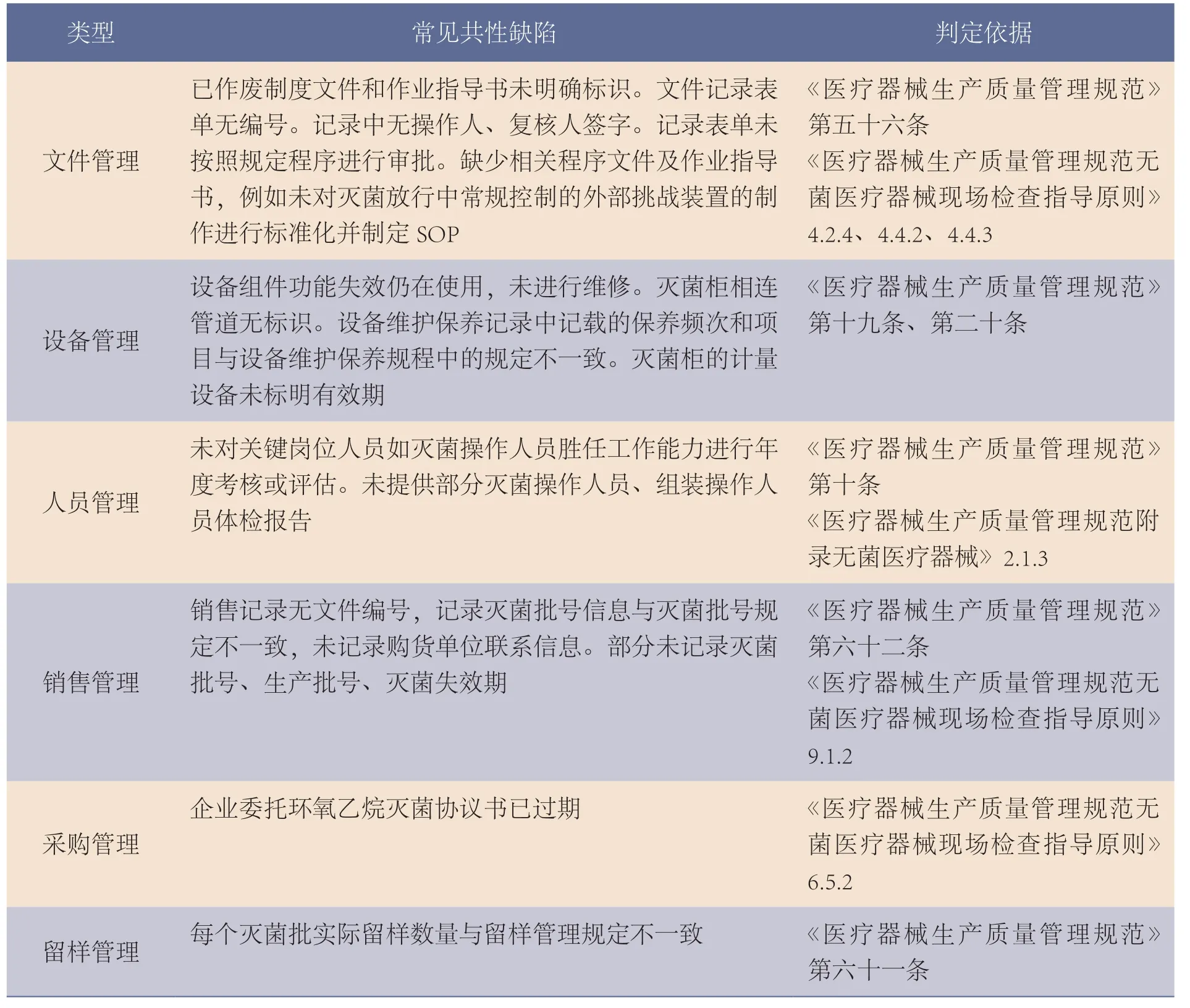
类型 常见共性缺陷 判定依据文件管理已作废制度文件和作业指导书未明确标识。文件记录表单无编号。记录中无操作人、复核人签字。记录表单未按照规定程序进行审批。缺少相关程序文件及作业指导书,例如未对灭菌放行中常规控制的外部挑战装置的制作进行标准化并制定SOP《医疗器械生产质量管理规范》第五十六条《医疗器械生产质量管理规范无菌医疗器械现场检查指导原则》4.2.4、4.4.2、4.4.3设备管理设备组件功能失效仍在使用,未进行维修。灭菌柜相连管道无标识。设备维护保养记录中记载的保养频次和项目与设备维护保养规程中的规定不一致。灭菌柜的计量设备未标明有效期《医疗器械生产质量管理规范》第十九条、第二十条人员管理未对关键岗位人员如灭菌操作人员胜任工作能力进行年度考核或评估。未提供部分灭菌操作人员、组装操作人员体检报告《医疗器械生产质量管理规范》第十条《医疗器械生产质量管理规范附录无菌医疗器械》2.1.3销售管理销售记录无文件编号,记录灭菌批号信息与灭菌批号规定不一致,未记录购货单位联系信息。部分未记录灭菌批号、生产批号、灭菌失效期《医疗器械生产质量管理规范》第六十二条《医疗器械生产质量管理规范无菌医疗器械现场检查指导原则》9.1.2企业委托环氧乙烷灭菌协议书已过期 《医疗器械生产质量管理规范无菌医疗器械现场检查指导原则》6.5.2留样管理 每个灭菌批实际留样数量与留样管理规定不一致 《医疗器械生产质量管理规范》第六十一条采购管理
进一步统计可看出,灭菌控制常见共性缺陷数量多达53种,其中以灭菌确认、仓储管理、灭菌过程控制3个方面的共性缺陷数量最多,占比分别为26%、19%和17%,三者合计占比达到了62%(图1)。一方面反映出生产企业对灭菌设备的系统设计、安装和验证方面经验的严重不足,过分依赖供应商;另一反面也反映出灭菌操作人员缺乏系统培训,在按照法规标准要求制定适合本企业产品的灭菌工艺文件、操作规程、检验规程,并严格按照文件要求进行记录方面意识薄弱,能省就省,成为灭菌安全性和有效性的隐患。
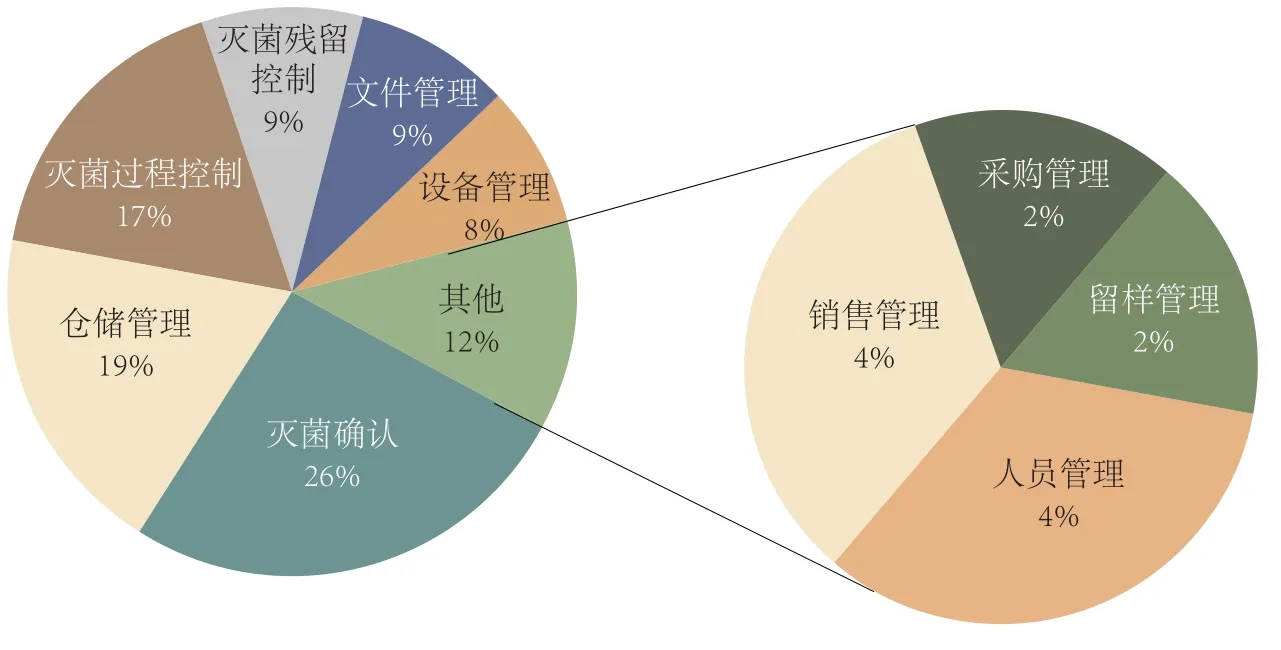
图1 环氧乙烷灭菌控制共性缺陷数量及占比分布图
2.3 灭菌控制GMP监管要点梳理
根据以上常见共性缺陷的分析,结合本人实际检查经验,现针对无菌医疗器械生产企业的灭菌控制环节,总结出以下7条监管要点:
(1)人员:灭菌控制相关岗位操作人员和检验人员是否经过专业技术培训,是否具备实际操作技能,是否按规定进行定期体检。
(2)设施:灭菌车间是否设置在专用房间内,是否有相应的安全、通风和排毒设施;EO存储区域是否独立和通风;解析是否在独立的柜室或房间内进行。
(3)设计开发:是否进行了灭菌确认,包括安装确认、运行确认和性能确认;是否对再确认启动的条件和周期进行了文件规定;灭菌确认报告中是否规定了灭菌参数和无菌保证水平、灭菌产品的呈现形式、产品的装载模式、解析的条件、产品的初始污染菌和微粒污染的可接受水平,微生物性能验证中是否对生物指示剂枯草杆菌芽孢放置分布进行了规定。
(4)文件控制:是否制定批号管理文件,并对生产批号和灭菌批号的组批方式进行规定;文件是否都有审核人和批准人签字,记录是否都有操作人员和复核人员签字。
(5)生产管理:灭菌记录中是否完整记载每批产品的灭菌参数(灭菌温度、相对湿度、EO暴露时间、EO浓度等)、有关设备编号、灭菌日期、操作人员与复核人员签名。
(6)质量控制:是否明确EO灭菌残留量的控制水平及检验方法,是否具备EO残留量的检验能力并按规定进行检验;灭菌设备上的计量器具、灭菌前后用到的检验设备是否按规定进行了定期校验并处于有效期内。
(7)销售:销售记录表单中是否完整记载产品名称、规格型号、数量、批号、有效期、购货单位名称、地址、联系方式。
以上要点基本覆盖了政府监管人员最常关注的现场检查切入点,通过这些切入点,监管人员可以更高效地发现灭菌控制环节的主要问题,并通过一系列溯源实现对整个生产质量管理体系的全面监督检查。
3.讨论
2018年是医疗器械GMP全面铺开的元年,也是医疗监管加强加严的关键之年。无菌医疗器械生产企业作为近几年医疗器械GMP“飞检”的重灾区,越来越受到关注。对于无菌医疗器械生产企业,灭菌控制往往是无法回避也是需要重点研究的系统工程。通过本文的分析和研究,我们从监管的角度梳理出了无菌医疗器械生产企业与灭菌控制有关的10大方面53种常见共性缺陷,并总结出7大监管要点。对于生产企业从业人员,通过对这些共性缺陷及其判定依据的详细了解,加上对以上监管要点的认真掌握,日常再配合法规标准的对照学习自查自纠,便可以快速提升企业的无菌产品生产质量管理水平。而对于政府监管人员,系统了解法规标准的判定准则,掌握灭菌控制常见共性缺陷的特征,也将有利于统一判定尺度,提升检查效率。
