Toxoplasma ROP16I/III ameliorated inflammatory bowel diseases via inducing M2 phenotype of macrophages
2019-12-16YongWeiXuRuiXinXingWenHuiZhangLuLiYiWuJingHuCongWangQingLiLuoJiLongShenXiChen
Yong-Wei Xu, Rui-Xin Xing, Wen-Hui Zhang, Lu Li, Yi Wu, Jing Hu, Cong Wang, Qing-Li Luo, Ji-Long Shen,Xi Chen
Abstract BACKGROUND Inflammatory bowel disease (IBD) is characterized by chronic and non-specific inflammation of the intestinal mucosa and mainly includes ulcerative colitis and Crohn's disease.AIM To explore the beneficial effect of ToxoROP16I/III-induced M2 phynotype macrophages in homeostasis of IBDs through downregulation of M1 inflammatory cells.METHODS RAW264.7 macrophages stimulated by lipopolysaccharide (LPS) (M1 cells) were co-cultured with Caco-2 cells as an inflammatory model of IBD in vitro. The expression of ToxoROP16I/III was observed in RAW264.7 macrophages that were transfected with pEGFP-rop16I/III. The phenotypes of M2 and M1 macrophage cells were assessed by quantitative real-time reverse transcriptase polymerase chain reaction and the expression of tumor necrosis factor (TNF)-α, interleukin(IL)-1β, IL-6, transforming growth factor (TGF)-β1, IL-10, inducible nitric oxide synthase (iNOS), and arginase-1 (Arg-1) was detected. The expression of iNOS,Arg-1, signal transducer and activator of transcription 3 (Stat3), p-Stat3, Stat6, p-Stat6, programmed death ligand-2 (PD-L2), caspase-3, -8, and -9 was analyzed by Western blotting, and Griess assays were performed to detect nitric oxide (NO).TNF-α, IL-1β, IL-6, TGF-β1, and IL-10 expression in the supernatants was detected by enzyme-linked immunosorbent assay, and Caco-2 cell apoptosis was determined by flow cytometry after mixing M1 cells with M2 cells in a Caco-2 cell co-culture system.RESULTS M1 cells exhibited significantly increased production of iNOS, NO, TNF-α, IL-1β,and IL-6, while ToxoROP16I/III induced macrophage bias to M2 cells in vitro,showing increased expression of Arg-1, IL-10 and TGF-β1 and elevated production of p-Stat3 and p-Stat6. The mixed M1 and M2 cell culture induced by ToxoROP16I/III exhibited decreased production of NO and iNOS and upregulated expression of Arg-1 and PD-L2. Accordingly, Caco-2 cells became apoptotic, and apoptosis-associated proteins such as caspase-3, -8 and -9 were dampened during co-culture of M1 and M2 cells. Flow cytometry analysis showed that co-culture of M1 cells with Caco-2 cells facilitated the apoptosis of Caco-2 cells, but co-culture of M1 and M2 cells alleviated Caco-2 cell apoptosis.CONCLUSION ToxoROP16I/III-induced M2 macrophages inhibited apoptosis of Caco-2 cells caused by M1 macrophages. This finding may help gain a better understanding of the underlying mechanism and represent a promising therapeutic strategy for IBDs.
Key words:Toxoplasma ROP16I/III; Caco-2; Inflammatory bowel disease; Immunity;Classically activated macrophages; Alternatively activated macrophages
INTRODUCTION
Inflammatory bowel disease (IBD), which includes ulcerative colitis (UC) and Crohn's disease (CD), is a chronic and non-specific gastrointestinal tract inflammatory disease characterized by an unexplained etiology and pathogenesis[1]. With changes in living conditions, the incidence of IBD is increasing, and the disease has gained growing attention due to its substantial impacts on patient quality of life[2]. The etiology of this disease remains unknown, and knowledge of the incidence and relevant risk factors,including environmental, genetic and immune factors and the gut microbiota, is increasingly emphasized worldwide[3]. The intestinal mucosal epithelium is an important immune organ in the body, and dendritic cells (DCs), and macrophages play pivotal roles in the immune responses of the intestinal mucosal epithelia to harmful substances produced upon intestinal epithelium damage induced by diet and cell ageing/death[4]. Immune cell activation and mediation of inflammation are complex processes[5]. In the body, the activation of inflammation occurs within a controllable range but can lead to systemic immune disorders when it falls outside this range. During the process of inflammation, macrophages play a central role in cell polarization and many different immunopathological phenomena. Under normal circumstances, the intestinal dynamic balance is strictly regulated by the mucosal immune environment[6]. However, once this important immunological homeostasis is destroyed, antigens can cause uncontrolled chronic intraluminal inflammation, which may contribute to immune disorders[7]. Some harmful macromolecular substances activate DCs and macrophages to induce the differentiation of T cells into proinflammatory Th1 and Th17 effector cells in IBD patients[8]. The proinflammatory cytokines, such as interleukin (IL)-2, IL-12, and interferon (IFN)-γ, produced by activated Th1 cells stimulate macrophages to secrete large amounts of other proinflammatory cytokines, including tumor necrosis factor (TNF)-α, IL-1β, and IL-6[2]. These cytokines can promote the proliferation of effector Th1 and Th17 cells and the release of chemokines, which attract more inflammatory cells to the site of inflammation to further amplify the proinflammatory immune response.
Lipopolysaccharide (LPS) is an important proinflammatory substance in medical research because of its unique properties, and its use creates in vitro pathogenesis of inflammatory reactions caused by bacterial infection[9]. Macrophages have been identified as an important factor in the progression of tissue inflammation[10].Macrophages have two obvious hallmarks, plasticity and diversity[11], and activated macrophages have two main phenotypes: classically activated (M1) and alternatively activated (M2)[12,13]. Accumulating evidence has shown that macrophages with distinct phenotypes exert diverse effects on inflammation and tissue repair[14,15]. LPS and IFN-γ can activate M1 macrophages via the nuclear factor kappa-B (NF-κB) signalling pathway, producing the proinflammatory factors IL-1β, TNF-α, IL-6, IL-23, reactive oxygen species, nitric oxide (NO), and inducible nitric oxide synthase (iNOS)[16]. Thus,M1 macrophages lead to inflammation and are predominant in the early stage of inflammation[17]. The cytokines IL-4, IL-10, and IL-13 activate M2 macrophages that are capable of modulating the immune response[18].
A series of reports indicated that helminths (parasitic worms) can induce type 2 immune intestinal inflammatory responses by promoting the expansion of protective bacterial communities that inhibit proinflammatory bacterial taxa[19]. Helminth exposure tends to inhibit IL-17 and IFN-γ production and promote IL-4, IL-10, and transform growth factor (TGF)-β release, induce CD4+ T cell Foxp3 expression (Treg)and generate regulatory macrophages, DCs, and B cells[20]. Helminth infection can induce the host to evoke a Th2 immune response that alternatively activates macrophages (M2)[21]. Helminths may subsequently skew the adaptive immune response towards Th2 and Treg responses, which are suggested to suppress the damaging Th1 and Th17 effector cells responsible for maintaining intestinal inflammation[22]. Thus, parasites and parasite-derived molecules likely have therapeutic potential in the prevention or control of immune-mediated illnesses.
Toxoplasma gondii (T. gondii) is an obligatory intracellular apicomplexan parasite that is capable of infecting a wide range of warm-blooded animals, and humans, and has a complex life cycle and pathogenic mechanism[18]. T. gondii can be divided into three archetypical genotypes: types I, II and III[23]. The virulence of T. gondii strains is closely related to the polymorphism of effector molecules carried by different genotypes[24]. Such effectors mainly include rhoptry proteins, dense granule proteins,micronemes, and pyramidal neurons[25]. Approximately 80% of all T. gondii isolates collected from animals and humans in China are of the Chinese 1 dominant genotype[26]that possesses the homology of ROP16 of type I and III [Toxoplasma ROP16I/III(ToxoROP16I/III)][27]. Melo MB demonstrated that ToxoROP16I/III, which harbours a tyrosine/serine kinase domain, can phosphorylate and activate the transcription factors signal transducer and activator of transcription 3 (Stat3) and Stat6[28], promote the polarization of M2 cells[29], reduce the production of IL-12 and enhance the synthesis of arginase-1 (Arg-1), IL-10, TGF-β1, and IL-13. This finding strongly suggests that the Toxoplasma-derived molecular effector ToxoROP16I/IIImight have potential in ameliorating bowel inflammation featuring type 1 dominant pathology by driving intestinal epithelial macrophages to M2 polarization. Our in vitro study showed that RAW264.7 macrophages could be biased to acquire an M2-like phenotype by transfecting lentivirus (Lv) carrying ToxoROP16I/III, and Caco-2 cell apoptosis and its associated proteins, such as caspase-3, -8, and -9, and were notably inhibited as shown by flow cytometry examination and analysis of the supernatants of M1 cells co-cultured with ToxoROP16I/III-induced macrophages. Thus, this study aims to identify how ToxoROP16I/III-induced M2 macrophages dampen the M1-mediated apoptosis of Caco-2 cells, which may provide a novel strategy for IBD immunotherapy with parasite-derived effector molecules.
MATERIALS AND METHODS
Reagents
Foetal bovine serum (FBS) was obtained from Wisent (Montreal, QC, Canada).Dulbecco’s Modified Eagle’s Medium (DMEM), the Griess Reagent System for measuring nitrite, primary antibody dilution buffer were all purchased from Beyotime (Shanghai, China). Nitrocellulose membranes were provided by Millipore(Billerica, MA, United States). Specific signals were detected using an enhanced chemiluminescence (ECL) kit (Thermo Scientific Inc., Waltham, MA, United States).LPS was purchased from Sigma (St. Louis, MO, United States). The mouse monoclonal arginase-1 (Arg-1) antibody was purchased from Proteintech (Chicago,IL, United States). The rabbit monoclonal iNOS antibody was manufactured by Abcam (Cambridge, MA, United States), and the human monoclonal antibodies against caspase-3, -8, and -9 were purchased from Cell Signaling Technology (CST,Danvers, MA, United States). The rabbit monoclonal antibodies against Stat3, Stat6, p-Stat3, and p-Stat6 were obtained from eBioscience (San Diego CA, United States) and programmed death ligand-2 (PD-L2) was obtained from Santa Cruz Biotechnology(Dallas, TX, United States). An Annexin V-FITC/PI apoptosis detection kit was purchased from BD Biosciences (BD, San Diego, CA, United States). Enzyme-linked immunosorbent assay (ELISA) kits for TNF-α, IL-6, IL-10, TGF-β1, and IL-1β were obtained from CUnited StatesBIO (Wuhan, China). Primer synthesis was completed by Sangon Biotech (Shanghai, China). TRIzol reagent was purchased from Invitrogen Life Technologies (Carlsbad, CA, United States). The HRP-conjugated anti-rabbit and anti-mouse IgG secondary antibodies were purchased from Proteintech (Wuhan,China).
Recombinant lentivirus plasmids
The open reading frame encoding ToxoROP16I/III(2124 bp, ToxoDB.org) was amplified from the entire Wh3 tachyzoite RNA, inserted into the recombinant p-EGFP plasmid,and directionally cloned to create pEGFP-rop16I/III. The recombinant Lv vector (LvpEGFP-rop16I/III) contained penicillin/streptomycin resistance and a Flag tag (Gene Chem Co., Shanghai, China).
Cell culture
RAW264.7 macrophages and Caco-2 cells (human epithelial colorectal adenocarcinoma cells) were preserved in the laboratory. The macrophages and Caco-2 cells were cultured in Dulbecco's modified eagle medium (DMEM) supplemented with 10% FBS and penicillin-streptomycin solution. All cells were cultured at 37 °C with 5% CO2in a humidified atmosphere. The macrophage medium was changed every 1-2 d, and the Caco-2 cell culture medium was replaced every 2-3 d. When the cells became 80% to 90% confluent, they were passaged and frozen for storage. A cell normally resides in a spherical, detached and undifferentiated state, known as the initial cell state.th
Transfection of RAW264.7 cells with recombinant lentivirus
Establishment of sufficient cell growth before the start of the experiment was necessary. For the experiments, macrophages were seeded at a density of 2 × 106cells per cm2into 12-well plates, and cells in the LV-pEGFP-rop16I/III-infected group became outstretched and significantly differentiated within 8 h. After 24 h, the medium was changed according to the state of the cells. Within 24 to 48 h after infection, the cells began to emit fluorescence. Recombinant Lv plasmids harbouring the target vector or empty plasmid were stably transfected into macrophages to generate LV-rop16I/III-Mφ and LV-Mφ, respectively. Polybrene reagents were added to all of the transfected cells, according to the manufacturer’s instructions. Caco-2 cells were maintained in the same medium.
LPS-induced macrophage polarization to M1 cells
The day before the cells were treated, macrophages were cultured in 6-well plates at a density of 2 × 106cells/mL. After the cells adhered and no pseudopodia was observable, the cells were stimulated with 1.0 μg/mL LPS for 24 h. At the end of the culture period, the culture medium was collected for NO and cytokine assays. iNOS expression was determined by Western blotting and quantitative real-time reverse transcriptase polymerase chain reaction (qRT-PCR) analysis, while the TNF-α, IL-6,and IL-1β expression levels were evaluated with qRT-PCR and ELISA. All of the above proteins were detected according to the manufacturer’s instructions. The results are presented as the mean ± standard deviation of three replicates from one representative experiment.
Cell co-culture system: gut inflammation of an IBD in vitro
To construct an inflammatory of IBD in vitro, we used a co-culture system comprising macrophages and Caco-2 cells seeded in the same well of transwell diverticulum[30-32].The macrophages were divided into five groups: M0 cells (control RAW264.7), M1 cells, LV-Mφ, LV-rop16I/III-Mφ, and mixed M1 and M2 cells. The transwell plates were seeded with macrophage cells at a density of 2 × 106cells per well and subsequently activated by 1.0 μg/mL LPS (apical side) for 6 h. These M1 macrophages, serving as the inflammation group, were transferred to polycarbonate membranes with a pore size of 0.4 μm (Corning, Corning, NY, United States).
For co-cultures with Caco-2 cells (basolateral), the cells were plated at a density of 5× 105cells. After 6 h, the M1 cell medium was replaced with fresh complete medium to avoid the effects of LPS on Caco-2 cells, and the Caco-2 cells co-cultures were then moved to transwell chambers for 24 h. Using the same method, M1 macrophages cocultured with M2 cells (upper side) were seeded in 6-well plates and then moved to transwell chambers containing Caco-2 cells.
NO assay
Macrophages, M1 cells, LV-Mφ, and LV-rop16I/III-Mφ cells were separately seeded in 6-well plates at 2 × 106cells per well. Macrophages and LV-rop16I/III-Mφ cells were separately seeded at 1 × 106cells per well and stimulated with 1.0 μg/mL LPS for 6 h.After 6 h, the medium was replaced with new complete medium. The M1 and M2 cell mixture was resuspended in 1 mL of common culture medium and cultured in 6-well plates at 37 °C and 5% CO2for 24 h. For the NO assay, culture medium (50 μL) was mixed with equal volumes (50 μL) of Griess reagent (I and II) in a 96-well plate and measured at an absorbance of 540 nm. A calibration standard curve was constructed,and the calculated concentration of nitrite dissolved in DMEM was calculated. All of the above experiments were performed according to the manufacturer’s instructions.
Western blotting analysis
Macrophages, M1 cells, LV-Mφ, LV-rop16I/III-Mφ, and mixed M1 and M2 cells were cultured in complete DMEM, and total protein was extracted after LPS stimulation for 6 h. The five groups listed above were also co-cultured with Caco-2 cells in transwell chambers. Then, the protein concentration was measured using a BCA protein assay kit (Beyotime, Shanghai, China). According to the assay results, the proteins were separated by standard 10% and 12% SDS polyacrylamide gel electrophoresis. Briefly,the proteins were electrotransferred onto nitrocellulose membranes, which were blocked with skim milk powder, washed with TBST 3 times for 10 min each,incubated with the corresponding primary antibodies, a horseradish peroxideconjugated secondary antibody and detected using an ECL kit. For detecting protein expression in all groups, the M1 marker iNOS (1:500), and the M2 marker Arg-1(1:1500) were used. Expression of the apoptotic proteins caspase-3 (1:2000), -8 (1:1500),and -9 (1:1500) was detected in all groups co-cultured with Caco-2 cells for 24 h.Importantly, LV-rop16I/III-Mφ cells exhibited anti-inflammatory factor protection by producing p-Stat3 (1:1500) and p-Stat6 (1:2000) via the activation of Stat3 (1:1000) and Stat6 (1:1000) signalling. Expression of the target proteins was normalized to that of the internal control mouse housekeeping gene encoding beta-actin (β-actin) (1:4000).HRP-conjugated anti-rabbit and anti-mouse (1:1000-10000) IgG served as the secondary antibodies.
mRNA extraction and qRT-PCR
Total RNA was extracted from the five groups of cells using TRIzol reagent. The ratio of absorbance at 260 nm and 280 nm was used to assess RNA purity. RNase-free,DNase-treated total RNA was reverse transcribed into cDNA using AMV reverse transcriptase. Real-time RT-PCR was performed with the Light Cycler 480 SYBR Green I Kit (Roche Diagnostics GmbH, Mannheim, Germany) using the gene-specific primers listed in Table 1. All of the experiments were performed following the manufacturer’s instructions. All amplification reactions were performed on a Light Cycler® 480 Instrument with an initial holding step (95 °C for 5 min) and 50 threestep PCR cycles (95 °C for 15 s, 60 °C for 15 s, 72 °C for 30 s). β-Actin was used as the normalization control for the evaluation of quantitative RT-PCR. Relative gene expression levels were determined using the 2-ΔΔCtmethod with Light Cycler 480 software (Roche, version 1.5.0).
ELISA
The five groups of cells were separately seeded in 6-well plates (2 × 106cells per well)and co-cultured with Caco-2 cells in 1 mL of complete culture medium on the apical and basolateral sides at 37 °C and 5% CO2for 24 h, and the cell supernatants were then collected. The inflammatory cytokines TNF-α, IL-1β, IL-6, IL-10, and TGF-β1were analyzed by ELISA in accordance with the manufacturer’s instructions. The absorbance was measured at 450 nm on an ELISA plate reader.
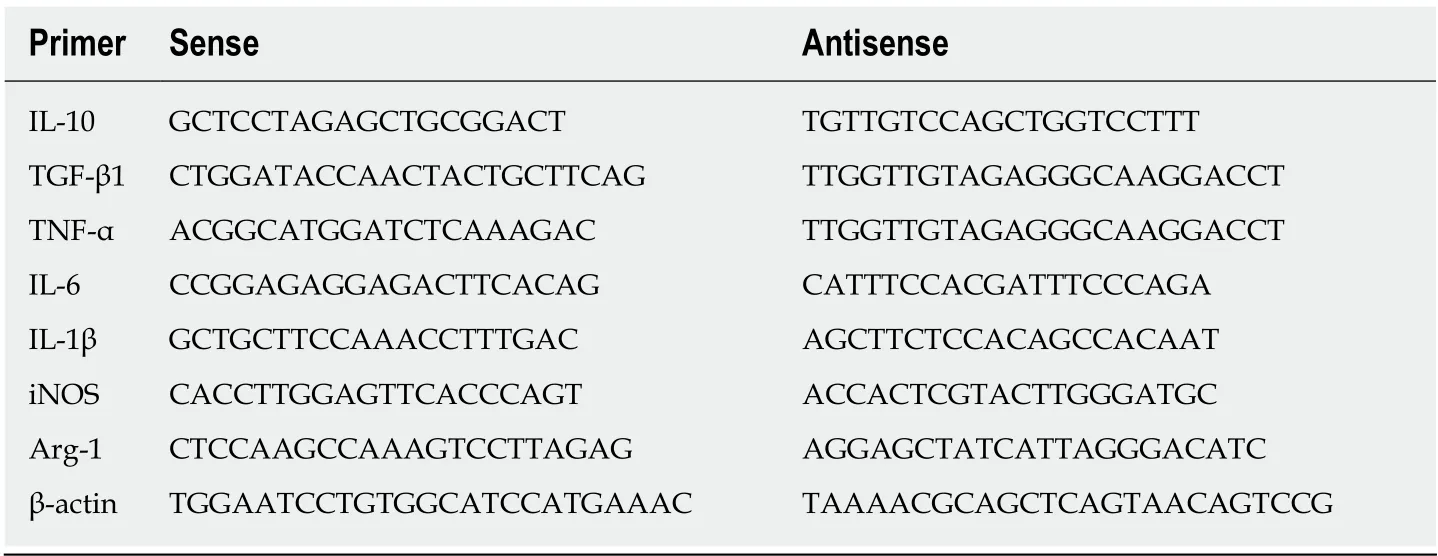
Table 1 The primers used for quantitative real-time reverse transcriptase polymerase chain reaction
Flow cytometry assay
After co-culture for 24 h, basolateral Caco-2 cells were collected and analyzed for apoptosis. The cells were washed once with cold PBS, and trypsin-EDTA solution was added to each culture group, followed by digestion using complete medium. All cells were washed three times and resuspended in binding buffer. Next, a blank tube containing neither FITC nor PI, a tube containing only 5 μL of Annexin V-FITC and a tube containing only 5 μL of PI were arranged separately. Other tubes contained 5 μL of Annexin V-FITC, 5 μL of PI and the resuspended cells. The cells were subsequently incubated for 15 min at room temperature in the dark and then analyzed by flow cytometry within 1 h. All of the above experiments were performed according to the manufacturer’s instructions.
Statistical analysis
All data are presented as the mean ± standard error of the mean (SEM). All experiments were replicated three times with similar results, and analysis was performed using GraphPad Prism software version 5.00 (GraphPad Software, San Diego, CA, United States). Comparison of the same parameters in multiple datasets or more than two groups was done using one-way analysis of variance with statistical significance at P < 0.05.
RESULTS
Macrophages stably transfected with LV-rop16I/III
LV-rop16I/IIIstably transfected macrophages were polarized to M2-like phenotype macrophages. Cells expressing recombinant pEGFP-Lv produced green fluorescence(Figure 1A). We detected the transfection of LV-rop16I/III-Mφ relative to LV-Mφ by Western blotting. The results showed that LV-rop16I/IIIwas successfully transferred into macrophages (Figure 1B and C).
LPS polarized RAW264.7 cells to the M1-like phenotype
To explore the optimal time for stimulation of macrophages with 1.0 μg/mL LPS, cells were seeded in 6-well plates at 2 × 106cells per well, and iNOS expression was detected at different time points over 24 h. The appropriate time point for cell and supernatant collection and detection was determined 6 h after LPS stimulation, when iNOS expression was detectable (Figure 2). We subsequently analyzed the relative mRNA expression of M1-like phenotype treated macrophages by qRT-PCR. IL-6(Figure 3A), IL-1β (Figure 3B), TNF-α (Figure 3C), and iNOS (Figure 3D) expression levels were markedly increased by LPS stimulation relative to normal macrophages.We measured the secretion of proinflammatory cytokines into cell supernatants by ELISA and found that the expression levels of IL-6 (Figure 3E), IL-1β (Figure 3F), and TNF-α (Figure 3G) were consistent with the relative mRNA results. In addition, the NO concentration (Figure 3H) and the iNOS (Figure 4F, H) protein expression were also significantly increased in the M1 inflammatory cell population relative to that in the other groups. No significant differences in any of the inflammatory cytokines
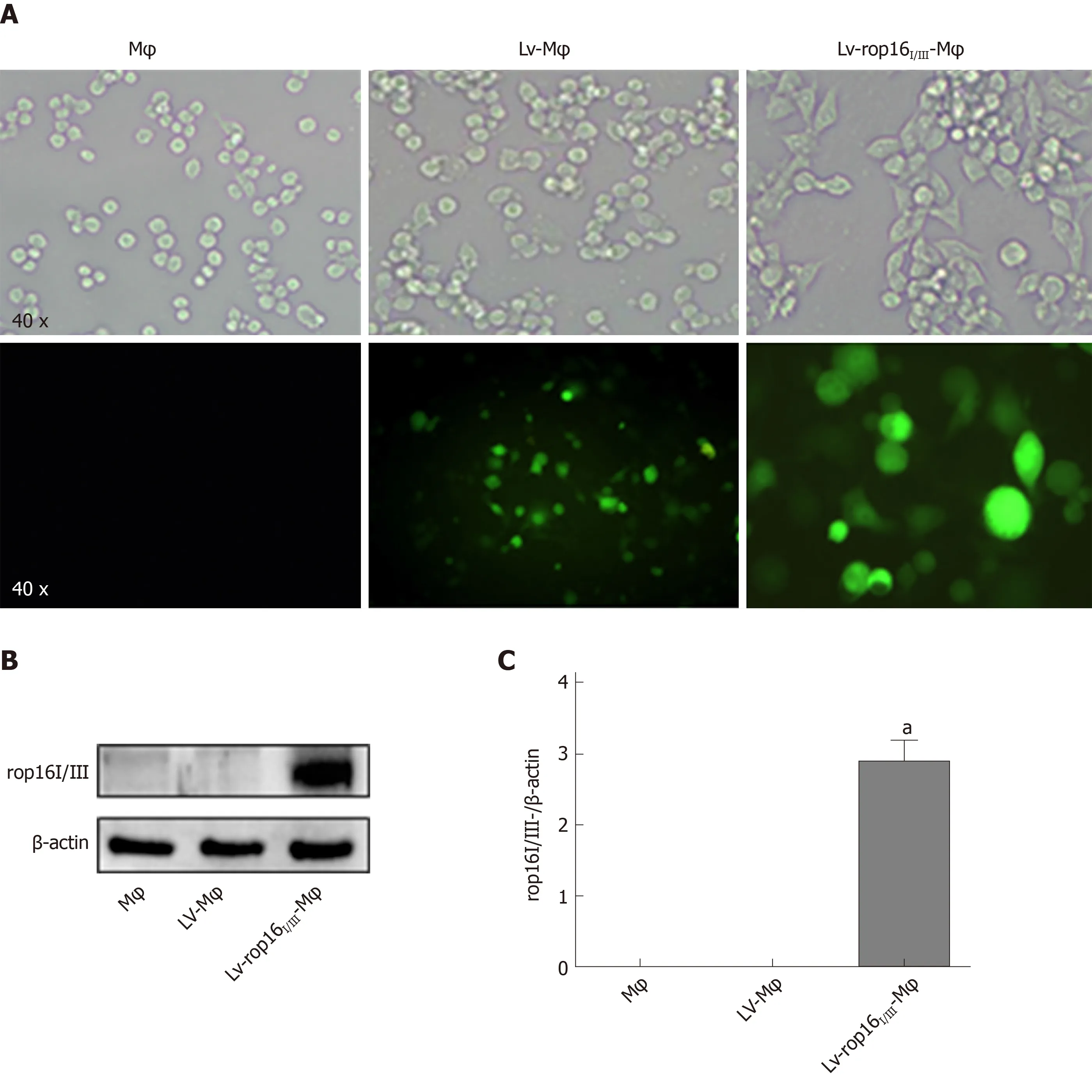
Figure 1 Stable transfection of RAW264.7 cells with LV-rop16I/III recombinant lentivirus.
Figure 1 stated above were found in the Lv- Mφ and Lv-rop16I/III-Mφ cells.
M2-like phenotype driven by LV-rop16I/III-Mφ
Compared with LV-Mφ cells, LV-rop16I/III-Mφ cells induced the M2-like phenotype in macrophages, which expressed high levels of activated (phosphorylated) Stat3 and Stat6 based on the detection of p-Stat3 (Figure 4A and B) and p-Stat6 (Figure 4A and 4C) by Western blotting. In addition, the protein expression levels of PD-L2 (Figure 4D and E) and Arg-1 (Figure 4F and G), which serve as M2-like phenotype markers,were increased, as determined by Western blotting. qRT-PCR was used to detect the mRNA expression of IL-10 (Figure 5A), TGF-β1 (Figure 5B), and Arg-1 (Figure 5C) in LV-rop16I/III-Mφ cells, and expression levels were significantly increased compared with that in LV-Mφ cells. The expression levels of IL-10 (Figure 5D) and TGF-β1(Figure 5E) were also sharply increased in stably transfected LV-rop16I/III-Mφ cell supernatants relative to LV-Mφ cell supernatants, as determined by ELISA, which was in accordance with the mRNA expression results.
M1 macrophages induced the apoptosis of Caco-2 cells in co-culture
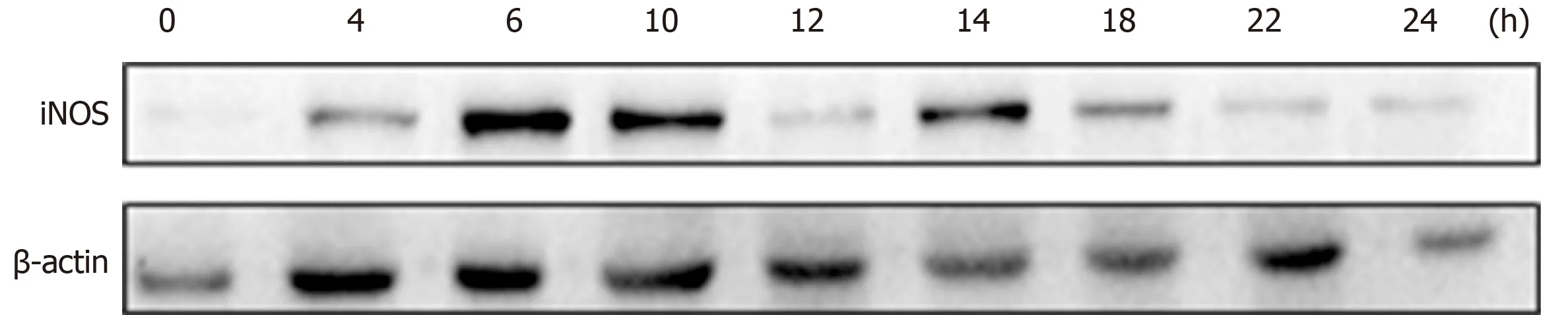
Figure 2 Lipopolysaccharide polarized to M1 cells.
LPS was used to polarize macrophages to the M1-like phenotype to produce the proinflammatory cytokines TNF-α, IL-6, and IL-1β, which could pass through 0.4-μm pore-size polycarbonate membranes. The LPS-stimulated macrophages were then cocultured with Caco-2 cells. The results showed that the protein expression of caspase-3 (Figure 6A and B), caspase-8 (Figure 6A and 6C), and caspase-9 (Figure 6A and 6D)was remarkably increased. Furthermore, the expression of apoptotic proteins in Caco-2 cells was significantly increased compared with that in normal macrophages, as determined by Western blotting. No significant difference in the expression of apoptotic proteins was noted between Lv-Mφ and LV-rop16I/III-Mφ cells. These results are in agreement with those from the flow cytometry assay (Figure 7). M1 macrophage-induced Caco-2 cell apoptosis was observed and compared with that observed during co-culture with Lv-Mφ and LV-rop16I/III-Mφ cells.
LV-rop16I/III-Mφ induced downregulation of M1 inflammatory cytokines
iNOS protein expression was evident after 6 h of LPS stimulation, and LV-rop16I/IIIMφ induced the polarization of macrophages to M2 cells. The medium was then removed, and M1 cells were mixed with M2 cells. Analysis of the relative mRNA expression in the M1 and M2 cell suspension by qRT-PCR showed that expression of the proinflammatory factors IL-6 (Figure 3A), IL-1β (Figure 3B) and TNF-α (Figure 3C) was remarkably downregulated in consistence with the ELISA results, and simultaneously, production of iNOS (Figure 3D) and NO (Figure 3H) was dampened compared with that in M1 cells. The caspase-3 (Figure 6A and B), caspase-8 (Figure 6A and C), and caspase-9 (Figure 6A and D) expression was markedly decreased,additionally, iNOS (Figure 8A and B) protein expression was reduced, Arg-1 (Figure 8A and C) and PD-L2 (Figure 8D and E) protein expression was stabilized, in the mixed M1 and M2 cell population compared with that in M1 cells, as determined by Western blotting. These results are consistent with those obtained from the flow cytometry assay (Figure 7). The co-culture of M1 cells with Caco-2 cells resulted in notably increased Caco-2 cell apoptosis and the expression of associated proteins relative to naive RAW264.7 macrophages. However, when M2 cells were added to the M1 suspension in the Caco-2 cell co-culture, Caco-2 cell apoptosis was remarkably ameliorated relative to M1 cells alone in co-culture (Figure 7A and B).
DISCUSSION
Previous investigations and our studies have demonstrated that helminth and helminth-derived products have the ability to suppress the development of IBD,mainly by downregulating Th1 and Th17 responses[33,34]. T. gondii, an intracellular parasite, has a diverse genetic structure. Type Chinese 1 (ToxoDB#9) is the dominant genotype in China according to recent investigations in both animals and human[26,35].Interestingly, recent studies have revealed that the rhoptry protein ROP16, secreted by type I/III Toxoplasma (ToxoROP16I/III) as a kinase, directly phosphorylates the Stat3/Stat6 transcription factors by bypassing the requirement for exogenous IL-4 and IL-13 and subverts host cytokine expression profiling during the early stage of innate immunity. ToxoROP16I/III-induced macrophages have features that resemble those of alternatively activated macrophages, termed M2 cells[24,36]. M2 cells are enriched during Th2 inflammation, such as that occurring during worm infections and asthma,because these immune responses are associated with IL-4 and IL-13 production,eosinophilia, and mucous production driven by Th2-polarized CD4 + T cell responses[37]. Additionally, M2 cells highly express arginase-1, TGF-β1, and IL-10,which have irreplaceable roles in suppressing excessive immune responses,particularly the Th1-dominant response. Thus, we assumed that the parasite-derived effector ToxoROP16I/IIImight have potential in ameliorating IBD (such as CD)pathology through downregulating the excessive Th1 and Th17 responses involved in the modulation of experimental pathogenesis of IBD in vitro. The present study aimed to investigate the therapeutic potential of ToxoROP16I/IIIas a new strategy in IBD immunotherapy.
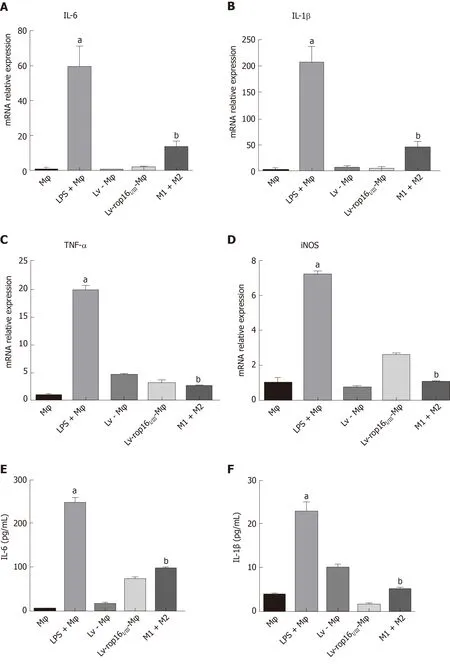
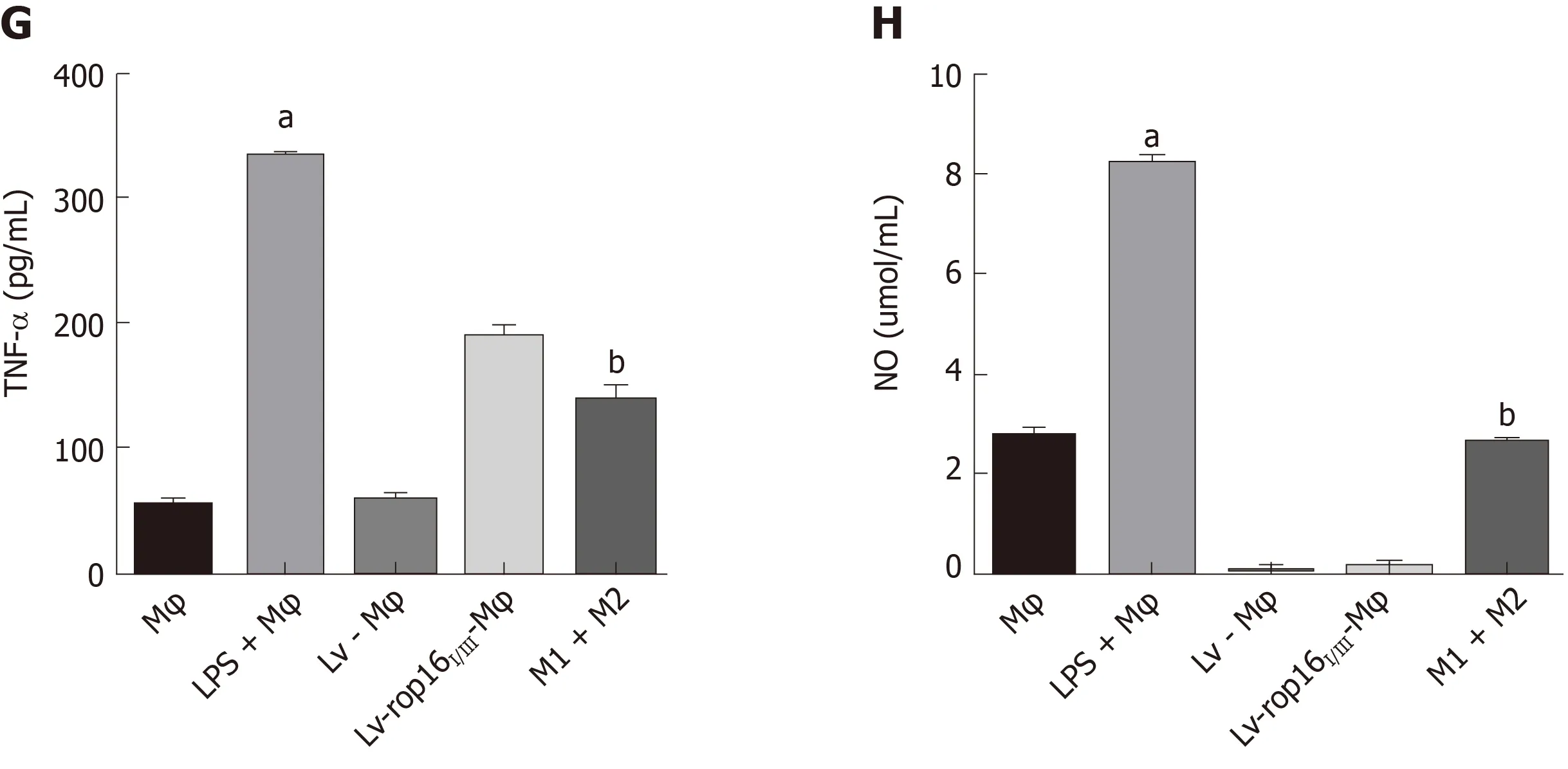
Figure 3 The proinflammatory cytokines produced by M1, and mixed M1 and M2 cells.
Known intestinal mucosal immune abnormalities, mucosal barrier defects, chronic infection, genetic and microbiota environments, and other factors have been associated with the pathogenesis of IBD[3]. In healthy intestinal mucosa, innate and adaptive immunity mechanisms control low-grade inflammation[38,39]. The intestinal microbiota is crucial for maintaining homeostasis of the intestinal tract and mucosa.When the balance or proportion of microbes is broken, or the bacteria become senescent and apoptotic, pyrolytic or macromolecular substances induce epithelial cell damage and produce innate immune and inflammatory reactions that drive the polarization of intestinal giant cells into M1 inflammatory cells and prompt the differentiation of original Th cells into Th1 cells.
The pathogenic basis underlying both CD and UC may be the dysregulation of normal immune responses in the intestinal mucosa[7]. In the local intestinal mucosal environment, secreted cytokines can activate macrophages, DCs, and neutrophils upon T cell activation[40]. In particular, Th1-dominant responses are thought to drive the pathogenesis of CD, while UC is driven by Th2 responses[41]. Activated macrophages are known to play a pivotal role in inducing the subsequent type 1 or type 2 response in adaptive immunity, which may extensively downregulate inflammatory reactions. Thus, immunomodulation may become a novel strategy of immunotherapy for the treatment of IBD.
Bacterial moieties, such as LPS and the Th1 cytokine IFN-γ, polarize macrophages towards the M1-like phenotype and promote the expression of numerous proinflammatory mediators. Therefore, we used the LPS-induced polarization of macrophages to M1 cells to activate the classical pathway and induce a Th1 immune response. The results paralleled those obtained from the ELISA cytokine assay with supernatants, Western blotting with cell protein and qRT-PCR. TNF-α, IL-6, and IL-1β proinflammatory cytokine production was remarkably increased. In contrast, M2 macrophages are associated with responses to anti-inflammatory reactions and tissue remodelling, as they express resistin-like-α (also known as Fizz1), Arg-1, chitinase 3,IL-10, and CD206[42-45].
Several earlier studies suggested that this co-culture model could imitate gut inflammation as seen in an IBD intestine in vitro. Tanoue et al[46]reported that established a gut inflammation in vitro model using intestinal epithelial cell line, Caco-2 cells and LPS stimulated-RAW264.7 cells. Kujawska et al[47], Wu et al[48], and Singh et al[49]and more experimental studies have used RAW264.7/Caco-2 to establish in vitro intestinal inflammation. According to previous research, we used LPS stimulated-RAW264.7 cells co-cultured with Caco-2 cells to establish an inflammatory of IBD in vitro.
Our data suggested that LPS induced the activation of NF-κB in macrophage cells and promoted the generation of proinflammatory M1 cells, in which iNOS, TNF-α, IL-1β, and IL-6 gene expression levels increased, while co-culture with Caco-2 cells in transwell plates increased the expression of the apoptotic proteins caspase-3, caspase-8, and caspase-9. In contrast, ROP16I/III-transfected macrophages showed phosphorylation and activation of the Stat3/Stat6 transcription factors and a distinctive profile of Arg-1, IL-10, and TGF-β1 expression that was consistent with the reported M2-like phenotype. These macrophages were mixed with M1 inflammatory cells, leading to the downregulation of inflammatory cytokines in M1 cells.
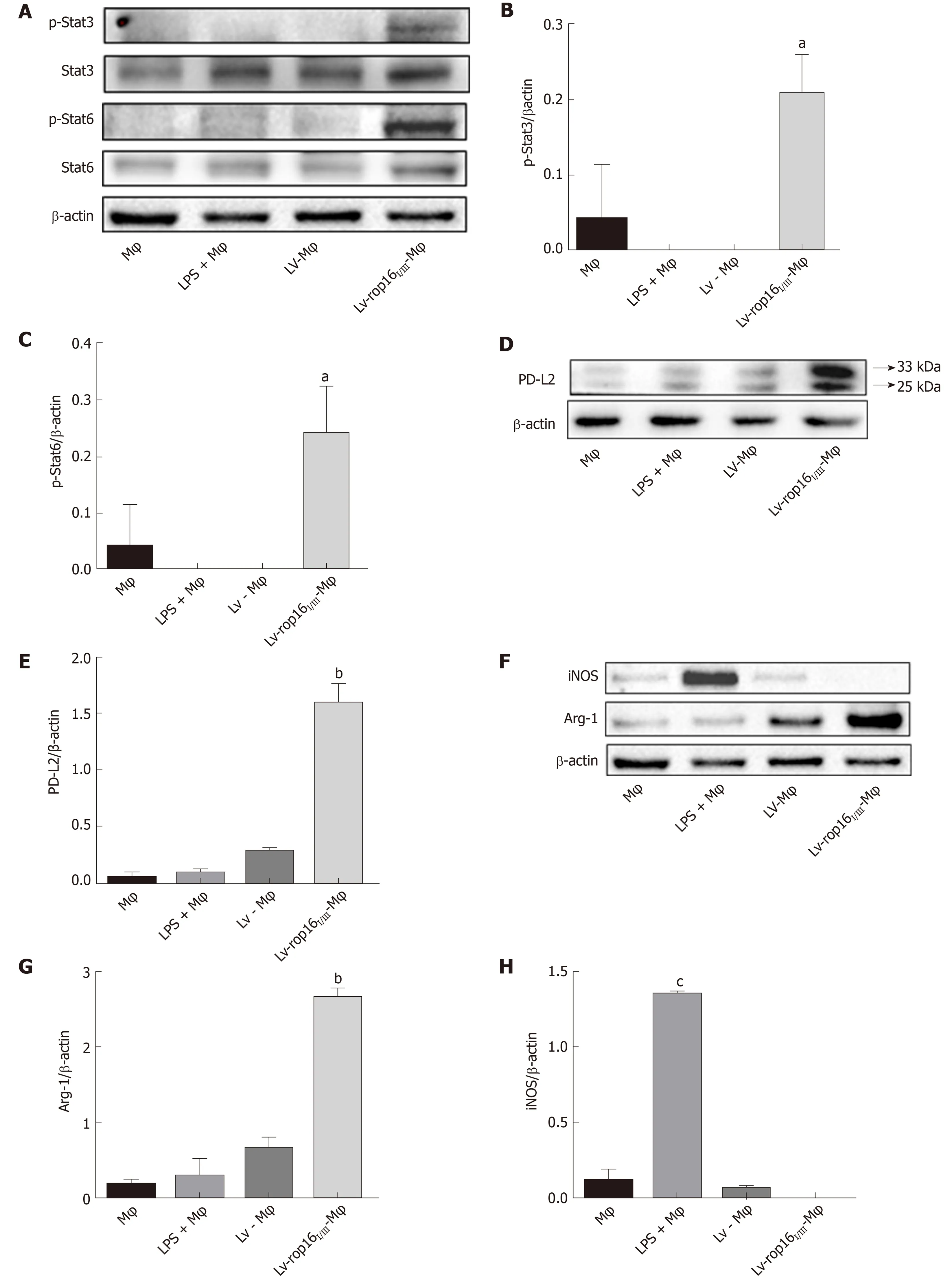
Figure 4 Western blotting analysis for the detection of M1 and M2 cell signatures.
M1 and M2 macrophages can be converted into each other in certain microenvironments, and the transformation of macrophages into different phenotypes regulates the initiation, development and cessation of inflammatory diseases[12,43]. In mixed M1 and M2 cell culture, expression of the proinflammatory cytokines TNF-α, IL-6, and IL-1β was remarkably downregulated when tested by qRT-PCR and ELISA. These cells were then co-cultured with Caco-2 cells, and the expression of apoptotic proteins decreased compared to that in M1 cells when detected by Western blotting and flow cytometry. M2 macrophages downregulated the expression of the inflammatory cytokines produced by M1 cells and hence inhibited the apoptosis of Caco-2 cells.
In this study, we presented additional evidence that the virulence-associated effector ToxoROP16I/IIImay induce the skewing of mouse M2 phenotype macrophages, altering cytokine profiles and prompting the differentiation of Th2 cells. Anti-inflammatory cytokines secreted by Th2 cells, such as TGF-β1, IL-4, IL-10,and IL-13, are involved in the humoural immune response, and the balance between Th1 and Th2 cells determines the balance between proinflammatory and antiinflammatory cytokines. In an ongoing study, novel in vivo approaches are being used to gain further insight into the potential role of ToxoROP16I/IIIfor IBD treatment.
Taken together, the experimental results presented herein demonstrated that the expression of NO, iNOS, TNF-α, IL-1β, and IL-6 by M1 cells, which generally accelerate the inflammatory process in IBD pathogenesis. Persistent secretion of the anti-inflammatory cytokines IL-10 and TGF-β1 by M2 cells in the local microenvironment can help maintain physiological status by downregulating the generation of proinflammatory factors, resulting in the alleviation of mucosal epithelium pathology in IBD, which may provide a novel strategy for IBD immunotherapy with parasite-derived effector molecules.
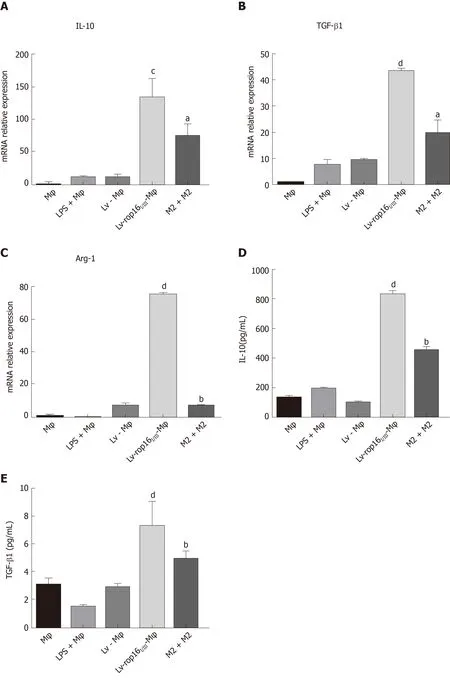
Figure 5 Cytokine expression was detected in M2 eclls and mixed M1 and M2 cells.
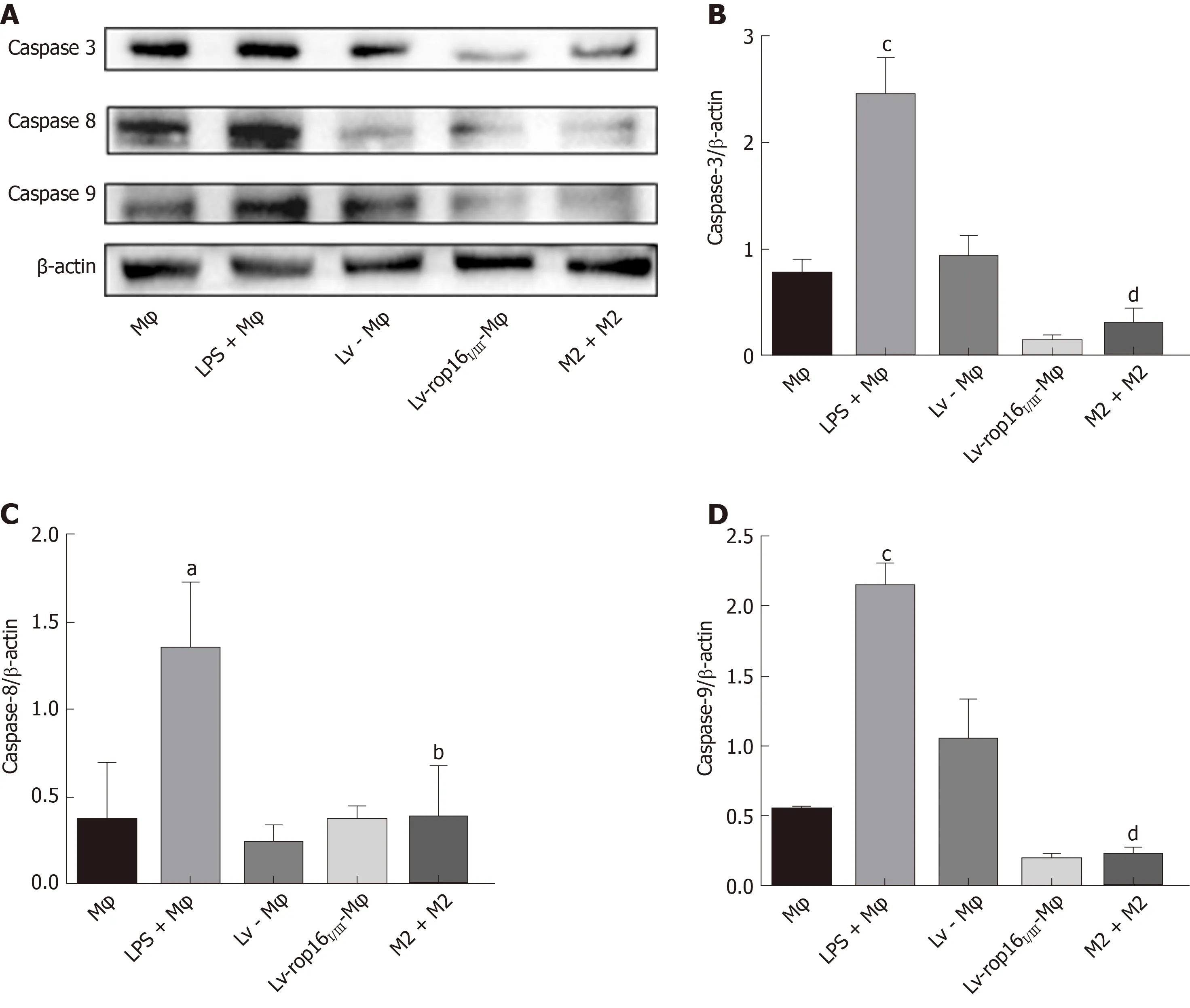
Figure 6 Caco-2 cell apoptosis was restrained by M1 cells mixed with M2 cells.
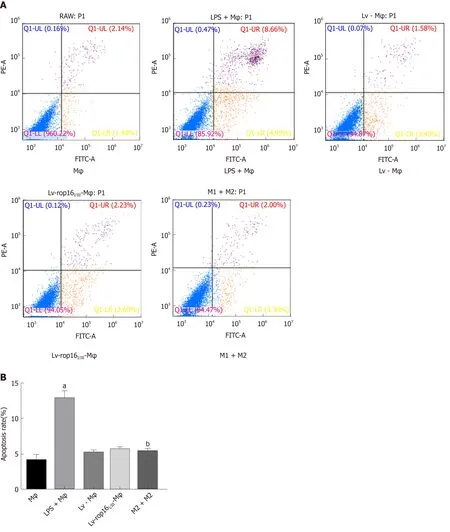
Figure 7 M1 cells mixed with M2 cells lead to reduction of Caco-2 cell apoptosis in co-culture.
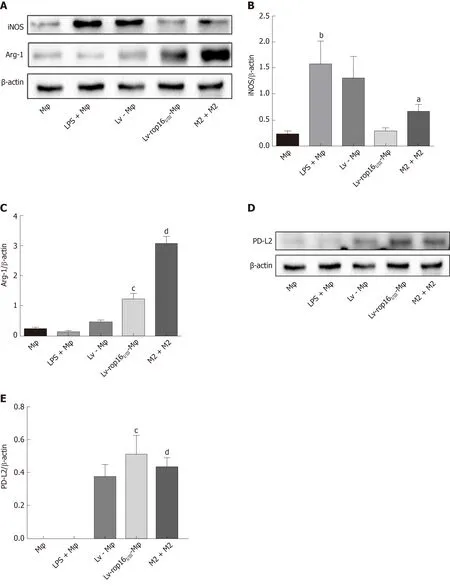
Figure 8 M2 cells reduced iNOS expression in M1 inflammatory macrophages.
ARTICLE HIGHLIGHTS
Research background
Inflammatory bowel disease (IBD) is characterized by chronic and non-specific inflammation of the intestinal mucosa and mainly includes ulcerative colitis and Crohn's disease. The incidence of IBD is increasing, and the disease has gained growing attention due to its substantial impacts on patient quality of life and increased side effects of traditional drugs in the treatment of IBD, so it is important to find new methods to treat IBD.
Research motivation
Toxoplasma ROP16I/III(ToxoROP16I/III) induced RAW264.7 polarization to M2 macrophage, downregulated the M1-associated inflammation response and played a protective role in Caco-2 intestinal epithelial cells.
Research objectives
The pathogenesis of IBDs remains unclear and the efficacy of current treatments is uncertain.Toxoplasma ROP16I/III-induced M2 macrophages might provide a promising strategy for the immunotherapy of IBDs using the parasite-derived molecules.
Research methods
ToxoROP16I/IIIinduced RAW264.7 polarization to M2 macrophage, enhanced the synthesis of arginase-1 (Arg-1), interleukin (IL)-10, transformed growth factor (TGF)-β1, and IL-13, downregulated the M1-associated inflammation response IL-1β, tumor necrosis factor (TNF)-α, IL-6,nitric oxide (NO), and inducible nitric oxide synthase (iNOS) as shown by quantitative real-time reverse transcriptase polymerase chain reaction. M1 and M2 cells co-cultured with Caco-2 cells through transwell alleviated Caco-2 cell apoptosis and its associated proteins by flow cytometry assay and Western blotting.
Research results
M1 cells exhibited dramatically increased production of iNOS, NO, TNF-α, IL-1β, and IL-6, while ToxoROP16I/IIIinduced macrophage bias to M2 cells in vitro, showing increased expression of Arg-1, IL-10, and TGF-β1 and elevated production of p-Stat3 and p-Stat6. The M2 mixed with M1 cell culture downregulated the production of iNOS, NO, TNF-α, IL-1β, and IL-6 by M1 cells,resulting in apoptotic alleviation of Caco-2 cells.
Research conclusions
ToxoROP16I/III-induced macrophages with an M2 phenotype inhibited the apoptosis of Caco-2 cells caused by lipopolysaccharide macrophage stimulation. These findings may be helpful for gaining a better understanding of the underlying mechanism and may represent a promising strategy for a novel immunotherapy against IBD.
Research perspectives
ToxoROP16I/IIImay be a new method for the treatment of IBD, and there are few side effects in the course of treatment. It will become another new aspect of study in the treatment of IBD.
ACKNOWLEDGEMENTS
We thank the Department of Pathogen Biology, Provincial Laboratories of Pathogen Biology and Zoonoses Anhui, Anhui Medical University for allowing this work to be performed there; and Prof. Chen Xi, Prof. Shen Jilong and Mr. Luo Qingli for their help in the experiment.
杂志排行

World Journal of Gastroenterology的其它文章
- Examining pathogenic concepts of autoimmune hepatitis for cues to future investigations and interventions
- LB100 ameliorates nonalcoholic fatty liver disease via the AMPK/Sirt1 pathway
- MicroRNA-760 acts as a tumor suppressor in gastric cancer development via inhibiting G-protein-coupled receptor kinase interacting protein-1 transcription
- Irisin attenuates intestinal injury, oxidative and endoplasmic reticulum stress in mice with L-arginine-induced acute pancreatitis
- Prognostic value of risk scoring systems for cirrhotic patients with variceal bleeding
- Gastrointestinal discomforts and dietary intake in Chinese urban elders: A cross-sectional study in eight cities of China