Examining pathogenic concepts of autoimmune hepatitis for cues to future investigations and interventions
2019-12-16AlbertCzaja
Albert J Czaja
Abstract BACKGROUND Multiple pathogenic mechanisms have been implicated in autoimmune hepatitis,but they have not fully explained susceptibility, triggering events, and maintenance or escalation of the disease. Furthermore, they have not identified a critical defect that can be targeted. The goals of this review are to examine the diverse pathogenic mechanisms that have been considered in autoimmune hepatitis, indicate investigational opportunities to validate their contribution, and suggest interventions that might evolve to modify their impact. English abstracts were identified in PubMed by multiple search terms. Full length articles were selected for review, and secondary and tertiary bibliographies were developed.Genetic and epigenetic factors can affect susceptibility by influencing the expression of immune regulatory genes. Thymic dysfunction, possibly related to deficient production of programmed cell death protein-1, can allow autoreactive T cells to escape deletion, and alterations in the intestinal microbiome may help overcome immune tolerance and affect gender bias. Environmental factors may trigger the disease or induce epigenetic changes in gene function. Molecular mimicry, epitope spread, bystander activation, neo-antigen production,lymphocytic polyspecificity, and disturbances in immune inhibitory mechanisms may maintain or escalate the disease. Interventions that modify epigenetic effects on gene expression, alter intestinal dysbiosis, eliminate deleterious environmental factors, and target critical pathogenic mechanisms are therapeutic possibilities that might reduce risk, individualize management, and improve outcome. In conclusion, diverse pathogenic mechanisms have been implicated in autoimmune hepatitis, and they may identify a critical factor or sequence that can be validated and used to direct future management and preventive strategies.
Key words: Autoimmune hepatitis; Pathogenesis; Epigenetics; Molecular mimicry;Epitope spread; Intestinal microbiome
INTRODUCTION
Autoimmune hepatitis is an immune-mediated chronic liver disease that lacks a demonstrable etiologic agent[1]. The diagnosis requires histological features of interface hepatitis, increased serum immunoglobulin G level, the presence of autoantibodies, and the exclusion of virus-related, drug-induced, metabolic, and hereditary diseases[1,2]. Type 1 autoimmune hepatitis is characterized by the presence of antinuclear antibodies and/or smooth muscle antibodies, and type 2 autoimmune hepatitis is characterized by the presence of antibodies to liver kidney microsome type 1 (anti-LKM1)[3,4]. Other autoantibodies that may be useful in the diagnosis of type 1 disease are antibodies to actin (anti-actin)[5-8], antibodies to soluble liver antigen[9,10],and atypical perinuclear anti-neutrophil cytoplasm antibodies[11,12]. Antibodies to liver cytosol type 1 may be useful in the diagnosis of type 2 disease[13-15].
Pathogenic concepts have indicated a genetic susceptibility for autoimmune hepatitis[16-18], and deficiencies or disruptions in homeostatic mechanisms have been described that can overcome self-tolerance[19-22]. These elements have not been formulated into a validated sequence that fully explains susceptibility, disease onset,and course. Furthermore, a critical pathogenic defect has not been identified that can be selectively targeted by a designed intervention.
A pathogenic hypothesis that can accommodate the clinical experiences and science of autoimmune hepatitis cannot exclude genetic makeup as a predisposing factor, but it must also integrate other factors to help explain the risk burden[19,23,24]. Epigenetic changes could translate environmental cues into alterations of gene expression that disrupt pathways modulating the immune response[25,26], and thymic failure may allow autoreactive T cells to escape negative selection[27-29]and help explain susceptibility to the disease.
Molecular mimicry could be a mechanism for foreign antigens (infectious agents,drugs, xenobiotic chemicals, and gut-derived bacteria) to overcome self-tolerance[30-33].Bystander activation could release pro-inflammatory cytokines, generate autoreactive T cells, and intensify the immune response[34-36]. Epitope spread could extend immune reactivity to less dominant epitopes within the same or other molecules[37-41]; neoantigens generated by tissue injury could expand the number of antigenic targets[34,42];and polyspecificity of the T cell antigen receptors (TCRs) could facilitate crossreactivity between foreign and self-antigens[43-45]. These mechanisms could help explain how environmental triggers can overcome immune tolerance and induce the disease. Furthermore, variations in the composition of the intestinal microbiome could allow microbial antigens and gut-driven immune cells to individualize the risk,clinical phenotype, and consequences of autoimmune hepatitis[46].
The goals of this review are to examine the diverse mechanisms that have been implicated in the susceptibility, onset, and maintenance of autoimmune hepatitis,indicate investigational opportunities to validate their pathogenic roles, and suggest future interventions that might modify their impact.
METHODS
English abstracts were identified in PubMed using the primary search words,“pathogenesis of autoimmune hepatitis”, “genetics of autoimmune hepatitis”,“epigenetics and autoimmunity”, “pathogens and autoimmune hepatitis”, “druginduced autoimmune hepatitis”, “molecular mimicry and autoimmune disease,”“neo-antigens”, “bystander activation”, “epitope spread”, “intestinal microbiome and autoimmune disease”, and “environment and autoimmunity”. Abstracts judged pertinent to the review were identified; key aspects were recorded; and full-length articles were selected from relevant abstracts. A secondary bibliography was developed from the references cited in the selected full-length articles, and additional PubMed searches were performed to expand the concepts developed in these articles.The discovery process was repeated, and a tertiary bibliography was developed after reviewing selected articles from the secondary bibliography. Over 1500 abstracts and 100 full length articles were reviewed.
FACTORS AFFECTING SUSCEPTIBILITY
Genetic predisposition
Genetic associations inside and outside the major histocompatibility complex (MHC)have been associated with the occurrence, serological phenotype, and severity of autoimmune hepatitis[18,47,48], and these associations have varied between ethnic groups[16,49-51], geographical regions[52-56], and age ranges[57,58](Table 1). Genetic factors within the MHC may predispose to autoimmune hepatitis by encoding antigen binding grooves on class II MHC molecules that select and present antigens with certain structural and conformational properties[17,59], and genetic factors outside the MHC (polymorphisms of key immune regulatory and cytokine-producing genes) may promote immune reactivity and inflammatory responses that affect clinical phenotype and disease severity[48,60-62](Figure 1).
The triggering peptides may be common in particular sub-populations, such as the young (viral infections) or the elderly (polypharmacy), and they may reflect the genetic composition and environment of the population at risk. HLA DRB1*03 has a low frequency in the normal Japanese population, and autoimmune hepatitis in Japan is associated mainly with HLA DRB1*04[49]. In contrast, HLA DRB1*04 is less frequent in the normal Italian population than in healthy North American adults (16% vs 34%,P = 0.0003)[63]. These differences in the genetic composition of the populations at risk could affect antigen selection, disease occurrence, and clinical phenotype.
Future studies of genetic and environmental factors associated with autoimmune hepatitis should be population-based and correlate genetic determinants with age,gender, ethnicity, and exposure to possible antigenic triggers[64]. Importantly, the riskburden for autoimmune hepatitis cannot be fully explained by genetic factors[23]. The key susceptibility alleles (DRB1*03:01 and DRB1*04:01) occur in only 51%-55% of white North American and northern European patients[16], and polymorphisms of genes outside the MHC, which have yet to be ascribed biological significance, have similar frequencies of occurrence[23,25,60,65,66]. Population-based genome-wide association studies may define the genetic fabric outside the MHC that is still undiscovered in autoimmune hepatitis, and epigenetic changes, cued by the environment, may also explain the risk.
Epigenetic changes
Epigenetic changes influence the function of genes without altering the sequence of deoxyribonucleic acid (DNA), and they may contribute to the occurrence and phenotype of autoimmune hepatitis[25,26](Table 1). They may also explain the riskburden for autoimmune hepatitis that cannot be linked to classical genetic associations. Epigenetic changes can occur at sites throughout the genome and respond to pressures that reflect diverse environmental factors[26,67](Figure 1). The structural adaptations occur within the nucleosomes of chromatin, and they affect the packaging of DNA, the activity of ribonucleic acid polymerase (RNAP), the ability of RNAP to open double-stranded DNA, and the accessibility of transcription factors to DNA binding sites[67-69]. DNA methylation[70]and histone modification[71,72]are the principal mechanisms that modify chromatin structure, and noncoding microribonucleic acids (miRNAs)[73-76]are the main agents that silence gene activity.
DNA methylation, histone modifications, and miRNAs
DNA methylation typically represses gene activity by inhibiting the binding of transcription factors to DNA, and it can be reversed by oxidation of the methylatedsite[26,77,78](Table 1). The ten eleven translocation oxygenases catalyze this conversion,increase gene translational activity, and counterbalance the repressive effects of DNA methylation[79-81]. Hypomethylation of gene promoters has been described in systemic lupus erythematosus (SLE)[82,83]and primary biliary cholangitis (PBC)[84], and epigenetic alterations in the expression of immune regulatory genes may promote loss of self-tolerance[85].

Table 1 Factors affecting susceptibility to autoimmune hepatitis
Histone modifications involve enzymatic alterations of amino acids in the tail of the histone proteins comprising the nucleosomes, and these modifications(phosphorylation, methylation, acetylation, and ubiquitination) can in turn loosen the wrap of DNA and increase the transcriptional activity of the gene[25,72,86,87](Table 1).Histone acetylation by histone acetyltransferase characterizes activated genes which may have stimulatory or repressive effects on disease activity by increasing the number and function of regulatory T cells (Tregs)[88]or increasing the expression of pro-inflammatory genes[89]. Histone deacetylase can modulate transcription activity by removing acetyl groups and repressing gene activity[90-92]. The number and location of possible histone modifications within the nucleosome contribute to genomic instability, and histone modifications have been associated with the loss of selftolerance[71,92,93].
Noncoding miRNAs are small double-stranded molecules that can pair with similarly sequenced messenger RNAs (mRNAs) within the cytosol. The complex is destined for degradation by a RNA-induced silencing complex[73,94-96](Table 1). Gene silencing can have organ specificity, and it can occur by degradation or repression of the mRNAs[97]. A single miRNA can affect the expression of multiple genes, and many different miRNAs can target the same mRNA. MiRNAs can thereby modulate disease activity by affecting immune (gene product production), inflammatory (cytokine production), and proliferative (lymphocyte differentiation) pathways[98-100]. The investigational challenge is to identify within the body of described miRNAs (> 1400 types) the key gene products that are targeted in autoimmune hepatitis[101,102].
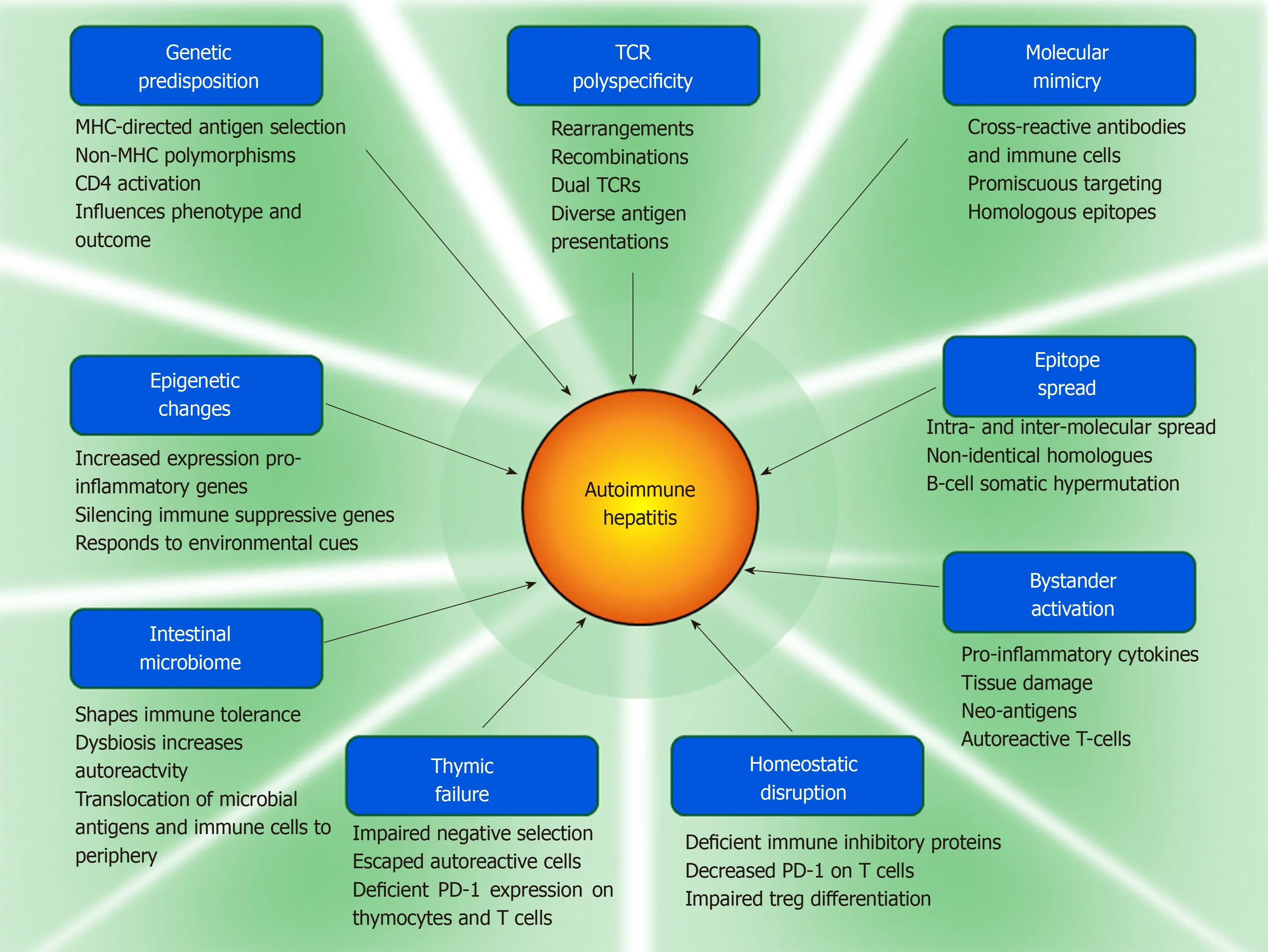
Figure 1 Pathogenic mechanisms implicated in autoimmune hepatitis.
Epigenetic changes and autoimmune hepatitis
Serum levels of micro-RNA 21 (miR-21) and micro-RNA 122 (miR-122) are increased in autoimmune hepatitis[103](Table 1). MiR-21 is expressed by T lymphocytes, and the programmed cell death 4 (PDCD4) gene is one of its gene targets[103,104]. The gene product of PDCD4 is the immune inhibitory protein, PDCD-4, and the downregulation of PDCD4 by miR-21 could promote immune reactivity. PDCD-4 increases the apoptosis of activated T lymphocytes and decreases the production of proinflammatory cytokines[105], and serum levels of miR-21 have correlated with the histological grade of liver inflammation[103].
Similarly, miR-122 has been associated with inflammatory activity (serum alanine aminotransferase levels) in autoimmune hepatitis[103], and miR-122 has increased the production of the pro-inflammatory cytokine, interferon type 1, by de-repressing cytokine signaling in a murine model of PBC[105]. Both miR-21 and miR-122 have been proposed as biomarkers of inflammatory activity in autoimmune hepatitis, but the nature and scope of their actions, disease-specificity, and value as therapeutic targets remain uncertain[103].
Thymic failure and escaped autoreactive T cells
Susceptibility to autoimmune disease may also relate to the escape of autoreactive immune cells from negative selection by the thymus and their persistence in the circulation (Figure 1). The existence of circulating autoreactive T cells implies a central defect in their thymic elimination or a peripheral failure to suppress their activity.Disruptions in the homeostatic molecular pathways that modulate negative selection within the thymus and lymphocyte differentiation in the lymphatic tissue have been proposed, and the expression of programmed cell death antigen-1 (PD-1) on thymocytes and maturing lymphocytes may be a key factor in limiting the escape and activity of autoreactive T cells[22].
Failures in thymic selection and peripheral protection
Immature CD4-CD8-thymocytes differentiate into CD4+CD8+thymocytes that are characterized by TCR beta (β)-chains (β-selection)[106-109](Table 1). The CD4+CD8+thymocytes with β-chains develop TCRs consisting of alpha (α)- and β-chains, and only the thymocytes that survive selection by demonstrating TCR specificity can mature into CD4+or CD8+T lymphocytes (repertoire selection)[110,111]. Thymocytes with self-reactive antigen receptors are eliminated by apoptosis (negative selection), and thymocytes that recognize foreign peptides presented by MHC molecules are selected to mature (positive selection)[27-29].
Self-reactive CD4+T cells that escape thymic deletion can autonomously express the immune inhibitory protein, PD-1, to induce self-tolerance[112](Table 1). Otherwise, the majority will persist as autoreactive T cells unless they express forkhead box P3(FoxP3)[112-114]. Self-reactive CD4+T cells expressing FoxP3 can differentiate into Tregs and counteract the autoreactive response. This transformation into Tregs occurs in only a minority of the self-reactive T cells that have evaded thymic elimination, and its dampening effect on the autoreactive response may be incomplete or negligible[112,113,115].
The expression of PD-1 on thymocytes[109,116]and maturing lymphocytes[112]is critical in limiting the emergence of autoreactive lymphocytes in the periphery. Clarification of the factors that regulate production and function of PD-1 should be an investigational objective in autoimmune hepatitis[22]. Soluble PD-1 is an alternatively spliced transcript encoded by the PD-1 gene which lacks a membrane-spanning portion and is rendered soluble[117]. The shortened soluble molecule is a variant of PD-1 that can competitively inhibit ligation of the full length molecule and prevent generation of an immune inhibitory signal[117-119]. Its counter effects on the immune inhibitory actions of full-length PD-1 may contribute to the escape of autoreactive T cells, and soluble PD-1 should be evaluated as a potential biomarker of disease activity and therapeutic target in autoimmune hepatitis.
TRIGGERING EVENTS
Microbial infection
Clinical experiences have described temporal associations between viral infections and the onset of autoimmune hepatitis in isolated cases. Infections with hepatitis A virus[120-125], hepatitis B virus (HBV)[126], hepatitis C virus (HCV)[127], Epstein-Barr virus(EBV)[128-132], varicella zoster[133], and human immunodeficiency virus[134]have been proposed as triggering events (Table 2). Other studies have described evidence of HCV[135-139], measles virus[140-142], cytomegalovirus (CMV)[143], and EBV[144]in the serum or liver tissue of patients with autoimmune hepatitis. These findings have supported an association between viral infection and autoimmune hepatitis, but the nature of the relationship (coincidental vs etiologic) has been uncertain[145,146].
The frequency of viral markers in patients with characteristic features of autoimmune hepatitis has been low (2%-11% for HCV and HBV)[136,147,148], often associated with false positive first generation assays confounded by hypergammaglobulinemia[149,150], and commonly unconfirmed by second generation assays[147](Table 2). The possibility of an undiscovered viral cause of autoimmune hepatitis cannot be excluded, but assessments for unusual viruses have been unrewarding (albeit few in number)[151,152]. After decades of observation and pursuit, a particular viral trigger for autoimmune hepatitis has not been established. Instead, viruses may constitute one of several categories of environmental agents that expose individuals to antigens that challenge immune tolerance in susceptible individuals.
The ubiquity and diversity of the presumed viral agents and the rarity of autoimmune hepatitis suggest that infection must be accompanied by host specific factors to trigger autoimmunity[147]. Genetic makeup[17,153], dysregulated critical homeostatic pathways[34], pre-conditioning by previous viral infections[154], molecular mimicries between microbial and self-antigens[30,31,155,156], and unmasked or newly created antigens (neo-antigens[34,146]or superantigens[157-159]) may constitute the hostspecific factors that translate microbial infection into an autoimmune disease.
Superantigens
Viruses and bacteria can induce the formation of superantigens which can activate autoreactive T cells without antigen presentation by MHC molecules[158,159](Table 2).The superantigens are a family of proteins that can bind to class II molecules of the MHC on antigen presenting cells (APCs) at a site distant from the peptide binding groove[158]. Simultaneously, they can bind to the TCR of T lymphocytes and activate them[158]. Superantigens bind to the variable region of the β chain within the TCR[160],and they frequently bind to several TCRs, thereby generating a polyclonal response[161]. Virtually all T cells bearing a particular TCR Vβ are activated by a superantigen[160], and the net effect is to trigger a T cell response manifested by the release of pro-inflammatory cytokines[162]and the proliferation of T lymphocytes[163,164].
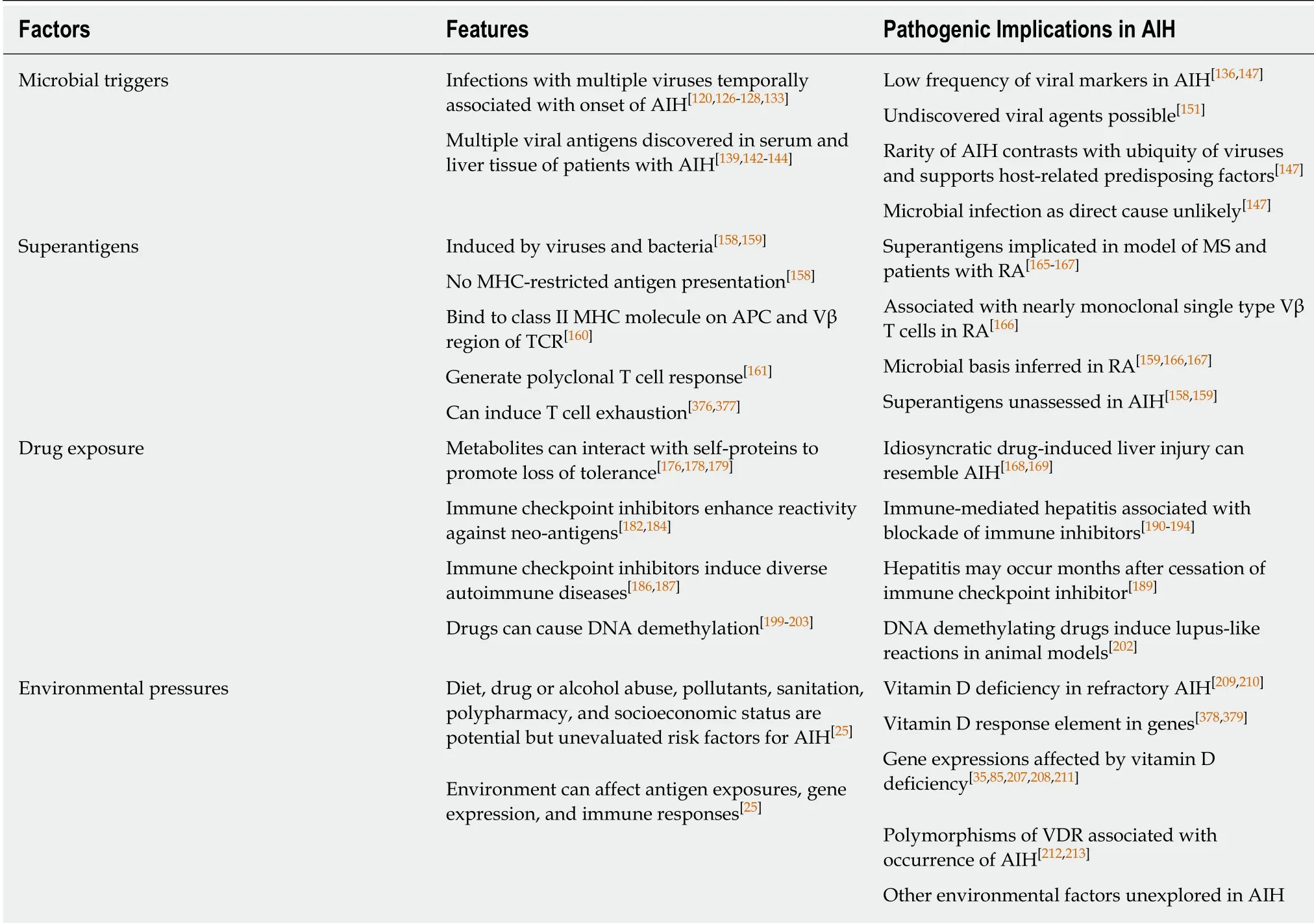
Table 2 Possible triggering events for autoimmune hepatitis
Superantigens have been implicated as a reactivating factor in an experimental model of multiple sclerosis[165]and a triggering factor in patients with rheumatoid arthritis[166,167](Table 2). The presence of a superantigen in patients with rheumatoid arthritis has been suspected because of the high percentage of Vβ14+T cells in the synovial fluid and the oligoclonality of the Vβ14+population[166]. Similar studies assessing the Vβ repertoire of T cells in the liver tissue and peripheral blood in autoimmune hepatitis might also implicate a superantigen and extend the search for microbial triggers.
Drug exposure
Multiple drugs, especially minocycline, nitrofurantoin, and infliximab, can induce an idiosyncratic liver injury indistinguishable from autoimmune hepatitis[168-170], and herbal medicines[171-174]and environmental pollutants[175]may also be initiating factors(Table 2). Drug-induced liver injury resembling autoimmune hepatitis is typically self-limited after drug withdrawal[168,169], and the rare instances of chronic selfperpetuating liver injury may reflect host-specific deficiencies in immune regulation and antigens generated by interactions between reactive drug metabolites and selfproteins that promote loss of self-tolerance[176-179].
The immune checkpoint inhibitors constitute an emerging category of biological agents that have been designed to block key immune inhibitory proteins [PD-1,programmed cell death antigen ligand-1, and cytotoxic T lymphocyte antigen-4(CTLA-4)][22,180,181](Table 2). The immune checkpoint inhibitors have enhanced the adaptive immune response to neo-antigens expressed by tumor cells in animal models and patients[182-185], and they have been associated with diverse immune-related adverse events[186-189], including the development of an immune-mediated hepatitis[190-194]. The clinical phenotype of liver injury induced by the immune checkpoint inhibitors has lacked the laboratory and histological features characteristic of autoimmune hepatitis[191,195-198], but the delayed development of hepatitis 8 mo after discontinuation of an anti-PD-1 preparation suggests that induced disturbances in the immune inhibitory axis may endure and pose a long-term risk for immune-mediated liver disease[189].
Drugs may also induce epigenetic changes that alter the expression of immune regulatory genes, and in turn this alteration that facilitate the loss of self-tolerance(Table 2). DNA demethylation has been associated with multiple drugs that can induce lupus-like reactions (procainamide, phenytoin, isoniazid, chlorpromazine,hydralazine, and 5-azacytidine)[199-203], and demethylating drugs as initiating agents in autoimmune hepatitis have been under-evaluated[25,169]. Aging may compound the effects of the environmental pressures on gene expression by promoting genomic instabilities through repeated cycles of DNA replication[204-206].
Most cases of autoimmune hepatitis lack a definable etiological trigger, and the possibilities that an antecedent microbial infection or drug exposure has been overlooked or that multiple different microbial and drug exposures have had a cumulative deleterious effect on the maintenance of self-tolerance cannot be proven or denied[146].
Environmental pressures
Epigenetic modifications can be induced by environmental pressures, and environmental cues that alter gene expression may help explain the occurrence of autoimmune hepatitis in diverse populations and geographical regions[35,85,207,208](Table 2). Vitamin D deficiency has been associated with histological severity, advanced hepatic fibrosis, and non-response to conventional glucocorticoid therapy in autoimmune hepatitis[209-211]. 1, 25 dihydroxyvitamin D binds to the vitamin D receptor(VDR), and this complex in turn activates the vitamin D response element in certain genes. This epigenetic change may enhance the transcription of genes that affect the inflammatory and immune responses[211], and genetic polymorphisms affecting the structure of the VDR have been associated with increased susceptibility to autoimmune hepatitis[212,213]. Other environmental pressures related to diet,socioeconomic status, drug or alcohol abuse, sanitation, and polypharmacy have an uncertain effect on the occurrence and outcome of autoimmune hepatitis, and they warrant further assessment in population-based studies.
FACTORS AFFECTING MAINTENANCE OR ESCALATION
Molecular mimicry
Molecular mimicry between foreign and self-antigens has been proposed as a mechanism by which the adaptive immune response can be sensitized to epitopes that are similar but not identical to self[30-33,214,215](Table 3). Invading pathogens[216-218],naturally occurring (environmental) antigens[35], synthetic peptides (chemical agents,halothane)[219-223], and vaccines[224,225]have been implicated as xenobiotic triggers that overcome self-tolerance by generating cross-reacting antibodies and immune cells(Figure 1). The molecular mimics have structural or conformational similarities with self-antigens, and they are introduced by infection, environmental exposure, or xenobiotic modification of proteins in situ[31,222,223].
Molecular identity rather than mimicry induces immune tolerance[226], and it is a mechanism by which invading pathogens may avoid recognition and elimination by immune defenses[227]. Molecular mimicry implies that the antigenic homologue is sufficiently different from the self-antigen to generate cross-reactive immune responses[31]. The molecular mimic must also resemble a self-antigen with biological activity that could promote autoimmune disease[31], and the structural or conformational similarities to the self-antigen must be sufficient to ensure binding to the same MHC class II molecules and activation of the same lymphocyte populations[228].
Pathogenic molecular mimicry has been incriminated in diverse autoimmune diseases that have been associated with bacterial and viral infections, including rheumatic fever[229-234], Guillain Barre syndrome[235,236], Lyme disease[237,238], Reiter’syndrome[239], ankylosing spondylitis[240,241], rheumatoid arthritis[242], multiple sclerosis[217], and SLE[243].
Molecular mimicry and autoimmune hepatitis
Molecular mimicries have been demonstrated between an autoantigen of autoimmune hepatitis (cytochrome P450 2D6) and infectious agents (Table 3). Theanti-LKM1 associated with type 2 autoimmune hepatitis recognize a short-linear amino acid sequence on cytochrome P450 2D6 as its principal epitope, and antibodies to herpes simplex type 1 virus, HCV, and CMV have demonstrated cross-recognition of this epitope[244-246]. Structural similarities between bacterial agents (Escherichia coli and Novoshingobium aromaticivorans) and pyruvate dehydrogenase-E2 complex (PDCE2) have also suggested a pathogenic role for molecular mimicry in PBC[247-251], and infection with a virus expressing a human P450 2D6 that is homologous to mouse P450 2D6 has induced severe liver damage in a murine model of autoimmune hepatitis[226,252]. The main investigational challenge is to associate these molecular mimicries with pertinent pathogenic mechanisms of the disease.
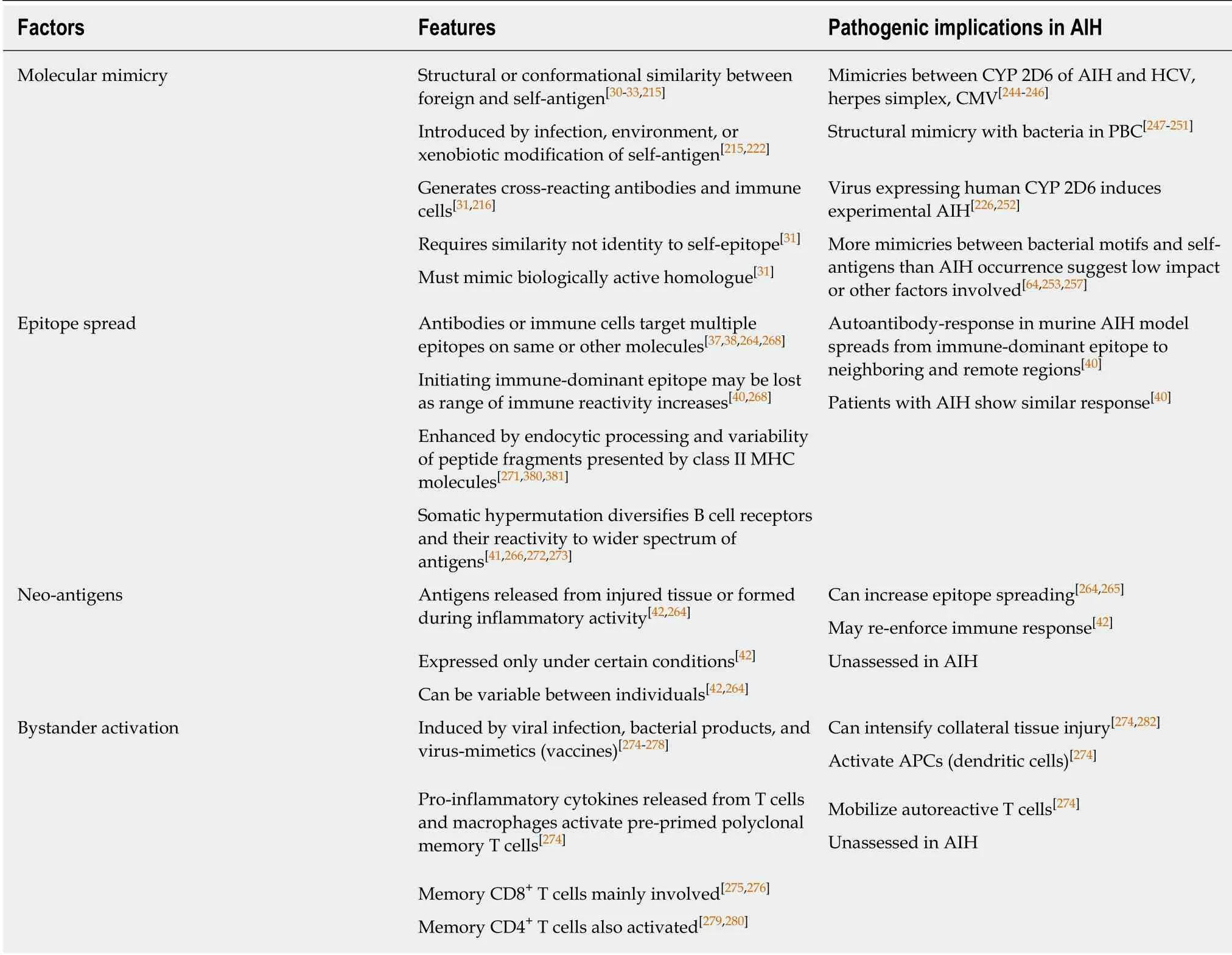
Table 3 Factors affecting maintenance or escalation of autoimmune hepatitis
Pathogenic uncertainties and future investigations
Studies analyzing exact peptide matches at penta-, hexa-, hepta-, and octapeptide levels have demonstrated that virtually all human proteins include a bacterial pentapeptide or hexapeptide motif[253](Table 3). Furthermore, the amino acid sequences necessary for peptide binding by MHC I and MHC II molecules are short(8-18 amino acids), and only a small portion of the antigen is required for recognition by a TCR[254-257]. The relative rarity of autoimmune diseases in the general population(5%-8%)[32]and low annual incidence of autoimmune hepatitis (0.85-1.9 cases per 100000 persons)[64]suggest that molecular mimicry is either an unimportant cause of autoimmune hepatitis or that its impact is mitigated by mechanisms that are still undiscovered.
Molecular mimicry induced by a non-infectious environmental agent has been uncommonly described in autoimmune disease, but it has been demonstrated in PBC.Immunization of a murine model with 2-octynoic acid, a chemical used widely in cosmetic products and food flavorings[221,258,259], has induced autoimmune cholangitis and antimitochondrial antibodies. The mechanism has been ascribed to a molecular mimicry in which the xenobiotic chemical has modified the lipoic binding region of PDC-E2 in situ and compromised immune tolerance of the self-antigen[222,223].
Future investigations in autoimmune hepatitis should explore molecular mimicries between non-infectious environmental agents and key antigenic targets in autoimmune hepatitis, such as cytochrome P450 2D6[244], formiminotransferase cyclodeaminase[14,15], and Sep [O-phosphoserine] tRNA: Sec [selenocysteine] tRNA synthase[260-263].
Epitope spread and neo-antigens
Autoantibodies and activated immune cells that target epitopes different from the immune-dominant epitope that initiated the immune response constitute epitope spread[37,38,40,41,264-266](Table 3). The reactivity may be against epitopes on the same molecule or homologous sequences on other molecules. Infections and environmental agents (chemicals, toxins, and drugs) may generate, amplify or sustain the autoimmune response by releasing neo-antigens directly from the injured tissue[37,38,264,267](Figure 1), and the neo-antigens may vary between individuals[42]. The autoantibodies or immune cells activated by the neo-antigens can broaden the range of immune reactivity and promote the spread of antigenic targets[37,41,268,269].
The initiating immune-dominant epitope may be lost during this process as the targeted homologous sequences in neighboring and remote regions become increasingly different from the original epitope[40]. In this fashion, the relationship to the initial immune-dominant epitope may be lost before clinical onset of the disease,and the original invading pathogen, environmental agent, or triggering antigen may be undetectable[270]. Epitope spread has been demonstrated in patients and animal models of autoimmune hepatitis characterized by antibodies to cytochrome P450 2D6[40].
Epitope spread can be enhanced by endocytic processing, variability in the peptide fragments that are loaded into the class II MHC molecules, and somatic hypermutation[35,41](Table 3). The peptide segments loaded into the class II MHC complexes after endocytic processing can vary in length and affinity[271]. Similar molecules can generate homologous peptide fragments that vary sufficiently to favor cross-reactive immune responses and thereby induce intermolecular epitope spread.Somatic hypermutation can also contribute to epitope spread by diversifying B cell receptors after antigen recognition in order to recognize a wider spectrum of foreign antigens[41,266,272](Table 3). Somatic hypermutation is an adaptive mutation involving single base substitutions mainly in the hypervariable regions of the immunoglobulin genes[272,273]. The roles of neo-antigens and somatic hypermutation in the severity of autoimmune hepatitis have not been assessed.
Bystander activation
Bystander activation can increase the immune response and tissue damage by activating pre-primed T cells and spreading the damage to uninvolved neighboring cells[274](Table 3). Viral infections[275], bacterial products (lipopolysaccharide)[276], and virus-mimetics (vaccines)[277,278]can induce the secretion of cytokines which in turn can stimulate the proliferation of polyclonal memory T cells. Memory CD8+T cells are mainly affected by bystander activation, and their proliferation is mediated by interferon-gamma and interleukin (IL)-12, IL-15, and IL-18[275,276,279,280]. Memory CD4+T cells can also undergo bystander activation (to a lesser degree), and their proliferation is mediated mainly by IL-2[279,280].
The bystander activation of pre-primed autoreactive T cells does not require stimulation of the TCR by a specific antigen[279,281]. Bystander activation with the release of pro-inflammatory cytokines can intensify the collateral damage associated with microbial infection[274,281-283], and it may be a mechanism that contributes to the emergence and progression of autoimmune disease by activating APCs and mobilizing autoreactive T cells[274,279](Figure 1). Bystander activation has not been assessed in autoimmune hepatitis, and the role of gut-derived microbial products in inducing a bystander effect has not been determined.
TCR polyspecificity
TCRs can undergo continuous rearrangements and re-combinations that broaden the range of antigens that they recognize, and this plasticity can contribute to an autoreactive response[284,285](Figure 1). TCRs recognize only a small portion of the peptide presented by class II MHC molecules[254,256,257], and the same TCR can respond to multiple antigens[45,286]. Polyspecificity of the TCR can generate cross-reactivity between T cells and facilitate promiscuous targeting of self-antigens.
T cell targeting can also be influenced by the surface expression of dual TCRs[287,288](Table 4). Thirty percent of human T cells express two functional α-chains. They may also express two β-chains at a lower frequency[43,289]. T cells expressing dual TCRs are more difficult to stimulate[44], and they may escape negative selection in the thymus by failing to demonstrate strong reactivity to self-antigens[290]. In the peripheral circulation, the T cells with dual TCRs may be stimulated by foreign antigen and selfpeptide, and the dual stimulation may overcome self-tolerance[44,288]. Future investigations in autoimmune hepatitis that determine the number and nature of circulating T lymphocytes with dual TCRs may reveal a means by which to measure or monitor the autoreactive propensity.
Intestinal dysbiosis
The intestinal microbiome shapes the intestinal and systemic immune responses[291-296](Figure 1). Its composition varies by gender, ethnicity, age, long-term diet, and socioeconomic status[297-300], and it constitutes an environmental variable that may impact on the predisposition, occurrence, and clinical phenotype of autoimmune disease. Bacterial components of the intestinal microbiome can activate Toll-like receptors (TLRs)[296,301-303], contribute to the formation of inflammasomes[304-307],stimulate the systemic immune response[293,296,308,309], and activate immune cells within the intestine that migrate to peripheral lymphoid tissue[310,311]. Changes in the microbial composition of the intestine (dysbiosis) have already been implicated in type 1 diabetes[312-314], rheumatoid arthritis[303,315-317], multiple sclerosis[318], inflammatory bowel disease[319-321], and diverse liver diseases, including NAFLD[306], PBC[249,322],PSC[323,324], and autoimmune hepatitis[325,326].
Intestinal microbiome and autoimmune hepatitis
Patients with autoimmune hepatitis have been distinguished from healthy individuals by deficiencies in the structural proteins (zona occludens 1 and occludin) that maintain integrity of the gastrointestinal mucosal barrier[325](Table 4). They also have had increased plasma levels of gut-derived bacterial lipopolysaccharide and decreased numbers of intestinal anaerobes (dysbiosis)[325]. Reduced diversity and total load of gut bacteria have also been associated with exacerbations of experimental autoimmune hepatitis in HLA DRB1*03-positive transgenic mice[326]. The findings suggest that autoimmune hepatitis is associated with dysbiosis, weakened gastrointestinal mucosal barrier, and translocation of gut-derived microbial products into the systemic circulation. The challenge is to establish the sequence of pathogenic events and distinguish causes from consequences of the liver disease.
Intestinal microbiome and gender bias
Autoimmune hepatitis occurs predominately in females, and this strong gender bias is evident in both children (where girls constitute 60%-76% of patients with autoimmune hepatitis) and adults (where women constitute 71%-95% of patients with autoimmune hepatitis)[64]. Explanations for the female propensity for autoimmune diseases have included estrogen effects on cytokine pathways[327]and gene expression[328], fetal microchimerism[329,330], and variable X-chromosomal inactivation[331]. The female bias for autoimmune disease may also be explained in part by gender-specific differences in the composition of the intestinal microbiome[300,332,333](Table 4).
Commensal microbes colonizing the intestine of male non-obese diabetic mice(NOD) can raise serum testosterone levels and protect them against the development of type 1 diabetes[300]. Transfer of the intestinal microbiota from mature male NOD mice to immature female NOD mice can protect the females from developing type 1 diabetes. Blockade of the androgen receptor can have a similar protective effect[300].Investigations of gender differences in the intestinal microbiome of patients with autoimmune hepatitis are needed to understand and possibly manipulate this pathogenic aspect.
Intestinal microbiome and the “hygiene hypothesis”
The “hygiene hypothesis” speculates that the rising risk of allergy (asthma, hay fever)and autoimmunity (type 1 diabetes, multiple sclerosis) in western countries relates to the decreasing incidence of infection and microbial exposure in childhood which in turn impairs development of the immune system[334-337]. Microbial exposure can promote maturation of the immune system by stimulating TLRs, inducing Tregs,increasing production of anti-inflammatory cytokines (e.g., IL-10), and generating an immune response that diminishes immune reactivity against self-antigens[334,338,339].
The intestinal microbiome has emerged as a key factor in maintaining immune tolerance by allowing systemic exposure to microbial products and activated immune cells during childhood and protecting against intestinal colonization by noncommensal micro-organisms[33,340](Table 4). Management strategies using extracts of bacterial and parasitic components have reduced the risk of type 1 diabetes in NOD mice, and the findings suggest that manipulations of the intestinal microbiome at ayoung age may protect against autoimmune disease[341,342]. The possibility that the intestinal microbiome can influence the risk, gender bias, severity, and outcome of autoimmune hepatitis is a compelling reason to investigate it further as a protective and pathogenic factor in this disease.

Table 4 Pathogenic implications of T cell antigen receptor polyspecificity and intestinal dysbiosis in autoimmune hepatitis
TRANSITIONING FROM PATHOGENIC CONCEPTS TO NEXT GENERATION MANAGEMENT
Translation of the pathogenic concepts into next generation management requires an understanding of each phase of autoimmune hepatitis from susceptibility to triggering events to mechanisms that sustain or intensify the inflammatory activity(Figure 2). The science of autoimmune hepatitis has not yet achieved this level of comprehension, but the opportunities to enlarge the knowledge base and impact on future management are plentiful.
Increased host susceptibility to an environmental factor is supported by genetic and epigenetic findings[16,18,25], and this susceptibility may be enhanced by defects in the negative selection of autoreactive immune cells within the thymus and alterations in the intestinal microbiome[27,46,112](Figure 2). Host susceptibility can be difficult to modify, but future investigations might validate mechanisms that ensure the expression of PD-1 on thymocytes and maturing lymphocytes[22], prevent or reverse DNA demethylation[25,343], or prime the intestinal microbiome to protect against autoreactive responses[341,342].
Progress has already been made in demonstrating that epigenetic manipulation is possible in the laboratory[344,345]. The methyl residue on S-adenosylmethionine can silence aberrant gene expression by inhibiting DNA demethylation directly or indirectly in cell culture[344]. The methyl groups donated by S-adenosylmethionine have a direct inhibitory effect on DNA demethylation, and they also impair the activity of the nuclear protein, methyl-DNA-binding domain protein 2 (MBD2). MBD2 binds specifically to methylated DNA, has demethylase activity[89,346,347], and is inversely associated with DNA hypo-methylation in patients with SLE[348].Investigational interventions that alter DNA demethylation directly or indirectly by methyl group donation[344]or the administration of anti-sense oligonucleotides that inhibit MBD2[345]have the prospect of dampening transcriptional activity of proinflammatory genes and reducing susceptibility for autoimmune hepatitis.
The identification of the triggering factors for autoimmune hepatitis is a key requirement for the development of protective strategies that might include environmental modifications or specific avoidance behaviors (Figure 2). Populationbased epidemiological studies that are designed to evaluate the association of infection, drugs, toxins, and xenobiotic chemicals with autoimmune hepatitis would

Figure 2 Pathogenic mechanisms and factors associated with susceptibility, onset, and maintenance of autoimmune hepatitis and possible therapeutic interventions.
Figure 2 help in identifying these triggers, and clarification of the role of the intestinal microbiome in affecting susceptibility, gender bias, and outcome could identify a modifiable source of microbial products and activated immune cells that might affect the occurrence and course of autoimmune hepatitis.
Multiple immune-mediated liver (NAFLD[306], PSC[323,324], and autoimmune hepatitis[325,326]) and non-liver (type 1 diabetes[313,314], rheumatoid arthritis[303,315,316],multiple sclerosis[318], and inflammatory bowel disease[319-321]) diseases have already been linked to intestinal dysbiosis, and modifications of the intestinal microbiome by diet, probiotics, antibiotics, or re-colonization are possible[46]. A preliminary randomized clinical trial of antibiotic therapy in PSC has suggested that therapy with vancomycin or metronidazole has the potential to improve liver tests and Mayo PSC risk score[349], presumably by favorably altering the intestinal microbiome[350].
Molecular mimicry, epitope spread, bystander activation, neo-antigen formation,TCR polyspecificity, and deficient immune inhibitory mechanisms (Tregs[351]and immune inhibitory proteins[22]) may extend an inciting event into a self-perpetuating autoimmune process (Figure 2). Multiple disturbances in the homeostatic pathways affecting activation[352], migration[353], and survival[354]of T lymphocytes have already been described that may be contributory. Deficiencies in the number and function of Tregs have been implicated[355,356](and challenged[357]), and failure of the immune inhibitory proteins, especially PD-1, to dampen the differentiation and proliferation of autoreactive cells has been an evolving area of investigational interest[22,358]. These disturbances may in turn relate to epigenetic changes that affect the transcription of immune regulatory genes, and these genes are already being manipulated experimentally[89,343,346].
Management strategies when the liver disease is established will depend on whether multiple factors must participate concurrently to sustain or escalate the disease or a predominant critical defect can be identified and targeted. Blanket immunosuppression may be necessary to suppress multiple concurrent pathogenic pathways, whereas highly specific interventions would require demonstration of a correctable pathogenic linchpin that supports the disease (Figure 2). Monoclonal antibodies against pro-inflammatory cytokines or B cells[359,360]and recombinant molecules that dampen T cell activation by increasing expression of the immune inhibitory proteins (recombinant CTLA-4[361-363]or PD-1[364]) are possible supplemental therapies. The adoptive transfer of antigen-specific, induced Tregs, mesenchymal stromal cells, or myeloid-derived suppressor cells exemplifies a next-generation intervention that might replace blanket immunosuppression[365,366].
CONCLUSION
Multiple pathogenic concepts have emerged to explain the susceptibility, onset, and maintenance of autoimmune hepatitis (Figure 1). Each concept is insufficient or incomplete. The risk burden for autoimmune hepatitis cannot be explained by genetic susceptibility; the triggering events of autoimmune hepatitis remain unknown; the factors that sustain or escalate the disease are unclear; and the dominant defect or particular aggregate of defects in immune homeostasis that could shape next generation therapy is unresolved.
Pathogenic concepts that are insufficient or incomplete are not wrong, and their deficiencies should drive investigations that define their impact more fully. The knowledge base of autoimmune hepatitis now includes evolving concepts of susceptibility, onset, and maintenance that should encourage investigations of acquired epigenetic change in gene expression, thymic dysfunction, molecular mimicry, epitope spread, and intestinal dysbiosis (Figure 2).
The next generation of management for autoimmune hepatitis will depend on clarification of the pathogenic sequence from susceptibility to established clinical phenotype, and it could include a strategy for prevention. Certain mechanisms may be evident throughout the course of the disease, and they may indicate a pathogenic linchpin that can be targeted selectively. Alternatively, certain mechanisms may require the alignment of several concurrent disruptions in immune homeostasis, and they may require blanket immunosuppression, possibly supplemented by site-specific interventions that target prominent contributory mechanisms. Studies that move the pathogenesis of autoimmune hepatitis closer to its cause may result in management strategies with a greater potential to replace blanket immunosuppressive regimens than interventions directed at downstream defects in the homeostatic network which are likely to yield supplemental rather than replacement interventions.
杂志排行

World Journal of Gastroenterology的其它文章
- LB100 ameliorates nonalcoholic fatty liver disease via the AMPK/Sirt1 pathway
- MicroRNA-760 acts as a tumor suppressor in gastric cancer development via inhibiting G-protein-coupled receptor kinase interacting protein-1 transcription
- Toxoplasma ROP16I/III ameliorated inflammatory bowel diseases via inducing M2 phenotype of macrophages
- Irisin attenuates intestinal injury, oxidative and endoplasmic reticulum stress in mice with L-arginine-induced acute pancreatitis
- Prognostic value of risk scoring systems for cirrhotic patients with variceal bleeding
- Gastrointestinal discomforts and dietary intake in Chinese urban elders: A cross-sectional study in eight cities of China