基于第一性原理的层状ZnO结构和化学键研究
2019-12-13苏亚拉
苏亚拉

摘 要:第一性原理系统的研究了层状ZnO的结构和化学键的特性。计算的结果表明层状ZnO既存在共价键又有离子键特性与其他人的结论相一致。
关键词:层状ZnO;几何结构;化学键
中图分类号:TB 文献标识码:A doi:10.19311/j.cnki.1672-3198.2019.34.089
0 引言
早在2010年,Pueyo等人就利用Zn4O4为前驱体成功地合成了层状六角结构的ZnO相。室温下,它的带隙能为~3.5eV,比纤锌矿ZnO略大。通过结构分析表明,这种层状ZnO的a-b面上Zn-O键长1.79,小于纤锌矿(B4)ZnO中Zn-O键长1.94。这种层状ZnO表现出与纤锌矿ZnO相近的光学带隙,有可能成为一种新型的光电子材料。Rakshit和Mahadevan首次通过第一性原理计算讨论了这种层状ZnO的稳定性。Molepo and Joubert预测这种层状ZnO可以通过纤锌矿ZnO高压相变获得,相变压力24.6GPa,但尚需进一步的实验验证。2015年,Wang等人利用超软赝势计算了这种层状ZnO的电子结构和光学性质。然而,对于层状ZnO物理特性的研究仍然没有深入的探讨,尤其是它的化学键。本文基于第一性原理密度泛函理论研究层状ZnO的化学键属性。
1 计算方法
基于密度泛函理论,计算采用模守恒赝势交换关联相互作用采用广义梯度近似(GGA)泛函。Zn和O的电子构型分别为3d104s2和2s22p4。能量优化采用 BFGS算法,平面波的截断能为900eV。B4相和层状Bk相ZnO的全布里渊区k点求和为7×7×4。测试结果表明进一步增加平面波的截断能和k点数目几乎对计算结果没有影响。
2 结果与讨论
2.1 结构与参数
为了评价计算结果的可靠性,我们首先几何优化了0GPa下B4相和层状ZnO的晶格常数(a,c,c/a)并与实验结果和他人的理论结果进行了比较,如表1所示。从表中可以看出,B4相和层状ZnO的计算结果与实验结果和他人的计算结果基本一致,基本参数a、c、c/a的计算误差小于2%,说明我们采用的计算方法和参数设置是可靠的。
2.2 分析化学键
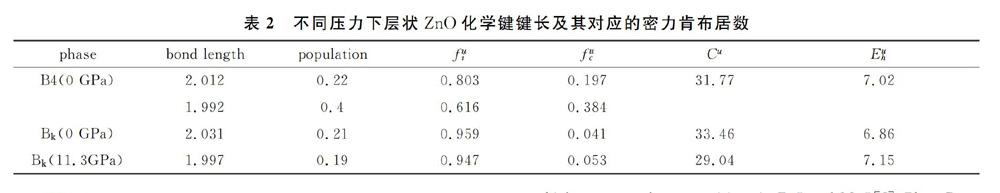
表1列出了0 GPa下B4相以及0 GPa和11.3GPa下层状ZnO化学键键长及其对应的密力肯布居数。0 GPa下B4相计算的键长与前人的结果基本相同。从表中可以看出,0 GPa下B4相的键长比0 GPa下层状ZnO化学键要长对应的密力肯布居数基本上没有变化。然而,在相转变压强下,层状ZnO化学键的键长减小。为了判断是否是离子键还是共价键,这可通过化学键理论分析。张等人提出过晶体的化学键理论,它是以化学键键能为基础的半经验方法。计算背景为:首先通过结构分析,将复杂的分子化学式分解成简单的二元分子式的组合,然后可以计算离子键和共价键的代表参数。对于ZnO本身就是最简单的二元分子式,如表2所示,其中代表键长。从表中数据可以看出,0 GPa下B4相计算得到化学键的异极化能大于同极化,这表明它是以离子性为主,与Phillips结论一致。但是我们计算的离子性标定为0.803大于Phillips的离子性标定0.616,这归因于计算的平均磁化率不同有关。对于0 GPa下层状ZnO,同样是以离子性为主。相转变后离子性标定减小,说明随着压强的增大离子性降低,這与我们以前的结论相同。通过计算得到不同化学键的离子性结果也能证明这一结论。总之,层状ZnO与B4相的ZnO一样,即表现出离子键又表现共价键特性。
3 结果
利用密度泛函理论研究了层状ZnO的几何结构参数和化学键的特性。根据化学键理论计算结果表明层状ZnO既存在共价键又有离子键特性。
参考文献
[1]Pueyo C L,Siroky S,Landsmann S,et al.Molecular precursor route to a metastable form of zinc oxide[J].Chem.Mater,2010,(22):4263.
[2]Rakshit B and Mahadevan P.Stability of the bulk phase of layered ZnO[J].Phys.Rev.Lett,2011,(107):085508.
[3]Molepo M P and Joubert D P.Computational study of the structural phases of ZnO[J].Phys .Rev.B,2011,(84):094110.
[4]Wang Q B,Zhou C,Wu J,et al.GGA+U study of the electronic and optical properties of hexagonal BN phase ZnO under pressure[J].Comp.Mater.Sci,2015,(102):196.
[5]Hamann D R,Schluter M,and Chiang C.Norm conserving pseudopotentials[J].J.Phys.Rev.Lett,1979,(43):1494.
[6]Perdew J P,Burke K,and Ernzerhof M.Generalized gradient approximation made simple[J].Phys.Rev.Lett,1996,(77):3865.
[7]Jaffe J E,Snyder J A,and Lin Z.LDA and GGA calculations for high-pressure phase transitions in ZnO and MgO[J].Phys.Rev.B,2000,(62):1660.
[8]Desgreniers S.Structural and compressive parameters high density phases of ZnO[J].Phys.Rev.B,1998,(58):14102-14105.
[9]Sun J and Wang H T.Ab initio investigations of optical properties of the high-pressure phases of ZnO[J].Phys Rev B,2005,(71):125132.
[10]Fan C,Wang Q,Li L X,et al. Bulk moduli of wurtzite,zinc-blende,and rocksalt phases of ZnO from chemical bond method and density functional theory[J].Appl.Phys.Lett,2008,(92):101917.
[11]Zhang S,Li H,Li L,et al. Calculation of bulk modulus on carbon nitrides with chemical bond method[J].Appl Phys Lett,2007,(91):251905.
[12]Phillips J C.Bonds and Bands in semiconductors Academic[Z].New York,1973.
[13]Vechten J A V.Quantum Dielectric Theory of Electronegativity in Covalent Systems.Electronic Dielectric Constant[J].Phys Rev,1969,(182):891.
[14]Serrano J,Romero A H,Manjon F J,et al. Pressure dependence of the lattice dynamics of ZnO:An ab initio approach[J].Phys.Rev.B,2004,(69):094306.
