基于Fe3O4过氧化物酶活性的Cd2+和Pb2+比色传感器
2019-12-11杨文平吴远根
杨文平,吴远根
1.贵州大学酿酒与食品工程学院,贵州省发酵工程与生物制药重点实验室,贵阳 550025;2.中国农业大学食品科学与营养工程学院,北京 100083
随着工业的高速发展,重金属污染越来越严重。环境中的重金属半衰期较长,难降解,易通过各种渠道进入食物链,富集在生物体内,引发多种疾病。例如,镉能引起肾小管功能障碍、中毒性肾损害和癌症等疾病[1-5]。铅可导致智力发育迟缓,尤其影响儿童大脑的发育[6-7]。鉴于其危害严重性,世界卫生组织、美国EPA和我国农业农村部均对环境和饮用水中的镉和铅重金属离子含量制定了严格的限量标准。目前,一些分析技术已普遍用于镉和铅的含量分析,如电感耦合等离子体质谱法[8-10]、电感耦合等离子体原子发射光谱法[11]、原子吸收/发射光谱法[12]、原子荧光光谱法[13]、反相高效液相色谱法[14]和电化学分析法[15]等。这些方法精确度和准确度较高,但一般需昂贵的检测仪器、专业的操作人员、复杂的样品前处理流程、高昂的检测成本以及较长的检测时间,不适合于发展中国家或偏远地区镉和铅重金属含量的实时在线监测。因此,为满足实际需要,亟需开发快速、简单和高效的镉和铅重金属离子检测方法。
近年来,许多新的快速检测方法已经建立,包括酶分析法、免疫分析法、试纸条法以及生物传感器法[16-20]。这些方法快速、简单、检测成本低,非常适合于偏远地区的镉和铅重金属离子的实时快速监测。但是,它们仍然存在一定的不足,例如:酶分析法所需的酶稳定性不高、易失活;免疫分析法需专门的抗原抗体且修饰过程相对复杂;试纸条法定量困难;电化学或荧光传感器法需专门的仪器。因此许多研究者将目光集中于比色传感器,其检测过程简单、检测结果肉眼可辨,但遗憾的是,基于纳米材料聚集显色的比色传感器易受非特异性靶标的干扰。因此,仍非常有必要借助其他技术弥补比色传感器的缺陷。
纳米酶是一类具有模拟酶特性的纳米材料[21],可催化底物氧化产生各种颜色变化。同时,纳米酶巨大的比表面积又非常有利于其表面的功能化。例如:Fe3O4具有过氧化物酶活性,可催化过氧化物酶底物氧化显色[22]。目前,基于Fe3O4催化活性建立的分析方法已成功应用于环境[23-25]、医学[26-28]和食品安全[29-30]等各个领域。在重金属检测方面,之前的研究大多集中于利用Fe3O4的吸附性能联合其他检测方法对重金属进行检测[31-32],而基于Fe3O4纳米酶直接催化的比色传感器较少。另外,核酸适配体是通过指数富集的配体进化技术(SELEX)筛选获得的对特定化学或生物靶物质具有高亲和力的DNA或RNA片段。其合成简单、易于修饰和具有靶向识别功能,已广泛应用于小分子、蛋白和细胞等各种物质的识别[33]。因此,本文联合具有催化活性的Fe3O4和具有一定特异性识别功能的核酸序列,建立了一种重金属比色检测方法。其中,检测体系的颜色与镉和铅浓度直接相关,可通过溶液颜色的变化或氧化产物特征吸收峰的强弱实现镉和铅离子浓度分析。
1 材料与方法
1.1 材料
1.1.1主要试剂 G4(序列:5′-GGGTGGGTGGGTGGGT-3′)由上海生工生物工程股份有限公司合成。六水氯化铁、乙二醇、尿素、聚乙二醇、4-氨基替比林及所有重金属标准品购自阿拉丁股份有限公司,其他试剂均为市售分析纯试剂。96-孔板购于康宁公司(Corning Incorporated)。实验用水均为二次蒸馏水。
1.1.2主要仪器 用全波长读数仪(Thermo Scientific Multiskan GO Microplate Spectrophotomer,USA)测量样品的紫外/可见吸收光谱和吸光值;通过扫描电镜(scanning electron microscope,SEM)观察磁珠的电子显微图像并测量其大小;用傅氏转换红外线光谱分析仪(FT-IR)获得红外光谱图;样品超声处理1 h后,用纳米粒径和zeta电位仪(Nano Particle &Zeta Potential Analyzer,USA)测量纳米粒的zeta电位。
1.2 方法
1.2.1检测原理 本文主要基于Fe3O4模拟酶催化活性及核酸的识别功能,建立了一种Cd2+和Pb2+比色检测法。Fe3O4具有过氧化物酶活性,可催化显色底物氧化产生颜色变化。如图1所示,当没有靶标离子存在时,核酸序列吸附于纳米粒表面,Fe3O4纳米酶对过氧化物酶底物催化活性较低,520 nm处产物特征吸收峰值较低。而当溶液中含有靶标重金属离子时,核酸序列可与Cd2+和Pb2+离子形成螯合笼结构[34-35],此复合物可增加纳米酶与底物之间的亲和力,同时核酸从Fe3O4上剥离下来,进一步促进Fe3O4氧化底物AAP产生肉眼可见的红色,产物特征吸收峰值较高。其中众多金属离子中,由于G4核酸序列与Cd2+和Pb2+离子的亲和力最强[36],所以在检测过程中仅含Cd2+和Pb2+离子的样品能显著增强Fe3O4的催化活性。因此,可以通过反应溶液颜色的变化和AAP产物特征吸收峰值的变化判断重金属是否超标。
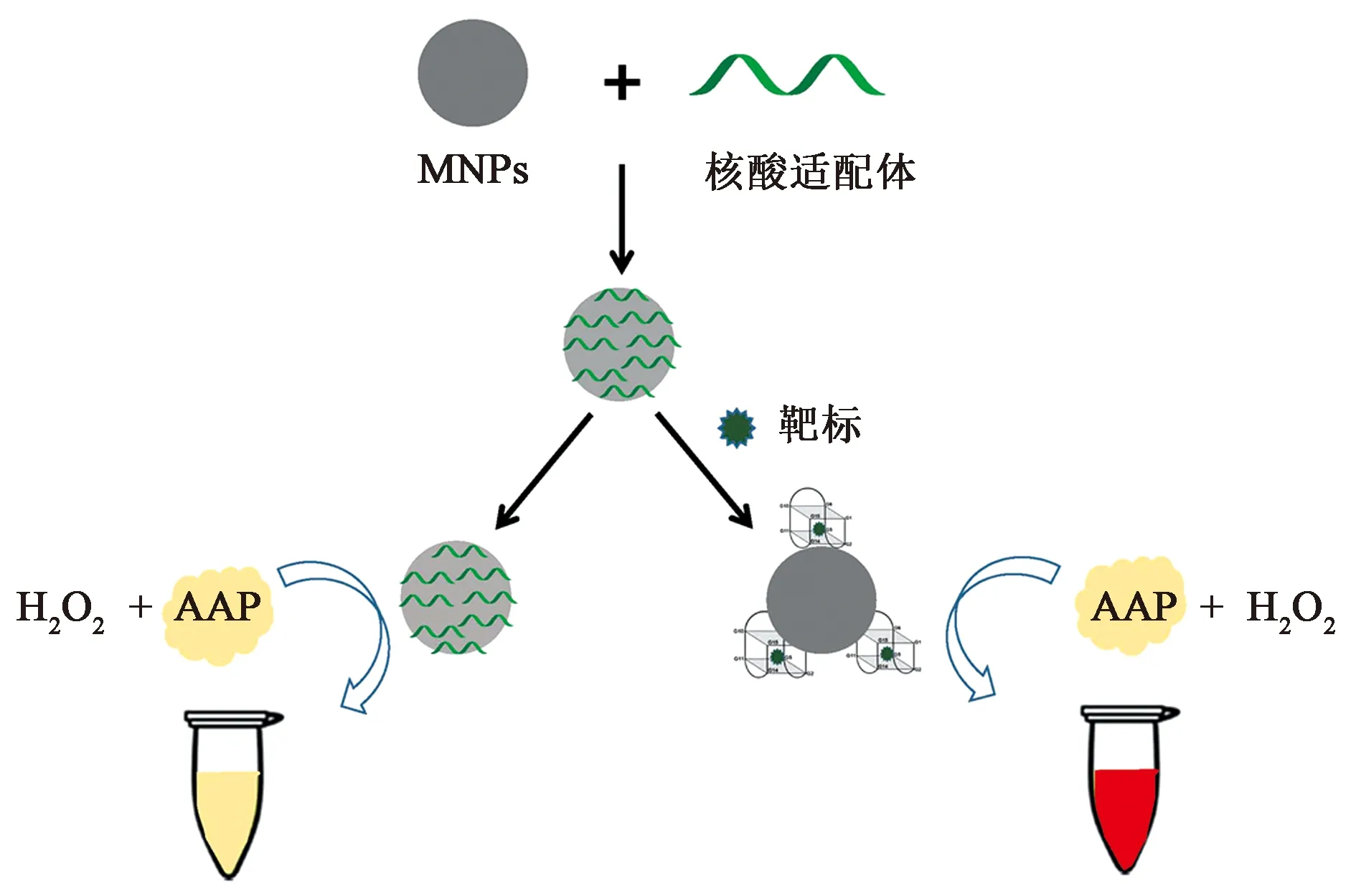
图1 基于Fe3O4过氧化物酶催化活性检测Cd2+和Pb2+重金属离子原理图Fig.1 Schematic illustration of the developed colorimetric sensors for Cd2+ and Pb2+ detection based on the peroxidase activity of Fe3O4
1.2.2磁珠的制备 参照文献[37]采用水热反应法制备磁珠。制备过程如下:称取1.15 g六水氯化铁溶于30 mL乙二醇中,混匀得到橘黄色液体。将2.0 g尿素和2.0 g聚乙二醇加至上述溶液中,磁力搅拌30 min形成均匀溶液。取28.8 mL上述溶液于反应釜中(约占反应釜容积80%),密封好于200 ℃下保温20 h。反应完毕,待反应釜冷却至室温,将产物转移至烧杯中,用蒸馏水和乙醇反复清洗,60 ℃干燥3 h得到黑色粉末。
1.2.3实验条件优化 本文以1 μmol/L靶标离子浓度为基准,分别考察缓冲溶液、缓冲溶液pH、底物、磁珠和核酸的浓度、反应时间等参数。最终,通过体系溶液520 nm波长处传感信号的强弱确定重金属检测的最优参数。
1.2.4性能测定 取5 μL 1 μmol/L G4和2 μL 靶标离子标准液于1.5 mL离心管中,空白对照加入等量二蒸水代替标准液,30 ℃孵育50 min。然后加入5 μL 3 mg/mL Fe3O4溶液,混匀后再孵育50 min。反应完毕,加入一定量的过氧化物酶底物(50 μL 500 mmol/L AAP和10 μL 0.5 mol/L H2O2)和428 μL 3.5 mmol/L NaAc-HAc缓冲溶液(pH 4.0)。取200 μL上述溶液进行紫外-可见吸光值测定。检测信号为ΔA520,其中ΔA=A(Cd2+)-A(空白)。特异性实验则通过向体系中添加等量(1 μmol/L)的竞争性重金属离子(Zn2+、Fe2+、Hg2+、Ag+、As3+、Al3+、Cu2+和Cr3+)测试此法特异性能。
2 结果与分析
2.1 可行性分析
如图2,单独的Fe3O4溶液在520 nm波长处无吸收峰(图2样1),仅含底物的反应体系520 nm波长处吸收峰较弱 (A为0.105)(图2样2),说明H2O2可诱导底物AAP发生轻微氧化。加入一定量Fe3O4纳米酶后,反应体系在520 nm波长处的吸光值迅速增加至0.223(图2样3),说明在含H2O2的条件下Fe3O4可催化底物AAP氧化成产物,且于520 nm波长处为产物特征吸收峰。当向体系中加入G4核酸时,反应体系520 nm波长处的吸光值有一定程度的下降(A为0.217),可能是由于核酸可以吸附在Fe3O4上,核酸的包封使纳米粒子与底物的接触受到了影响,从而降低其酶的催化活性。然而当有靶标Cd2+和Pb2+存在时,其520 nm波长处的吸收峰明显增加,说明Cd2+和Pb2+离子可与Fe3O4竞争性结合G4核酸,使核酸从Fe3O4剥离下来,或者形成的复合物能诱导Fe3O4的催化活性,增强其对底物AAP的氧化能力。
2.2 Fe3O4纳米粒子的表征
本文制备的磁性Fe3O4纳米粒子,SEM表征结果说明其形状为球形,粒径大小约20 nm(图3A)。FT-IR能谱表征结果如图3B,602 cm-1处的强吸收峰与Fe-O键相关,1 628 cm-1处的吸收峰为N-H剪式振动,1 090 cm-1和892 cm-1处的吸收峰为胺的N-H摇摆振动。
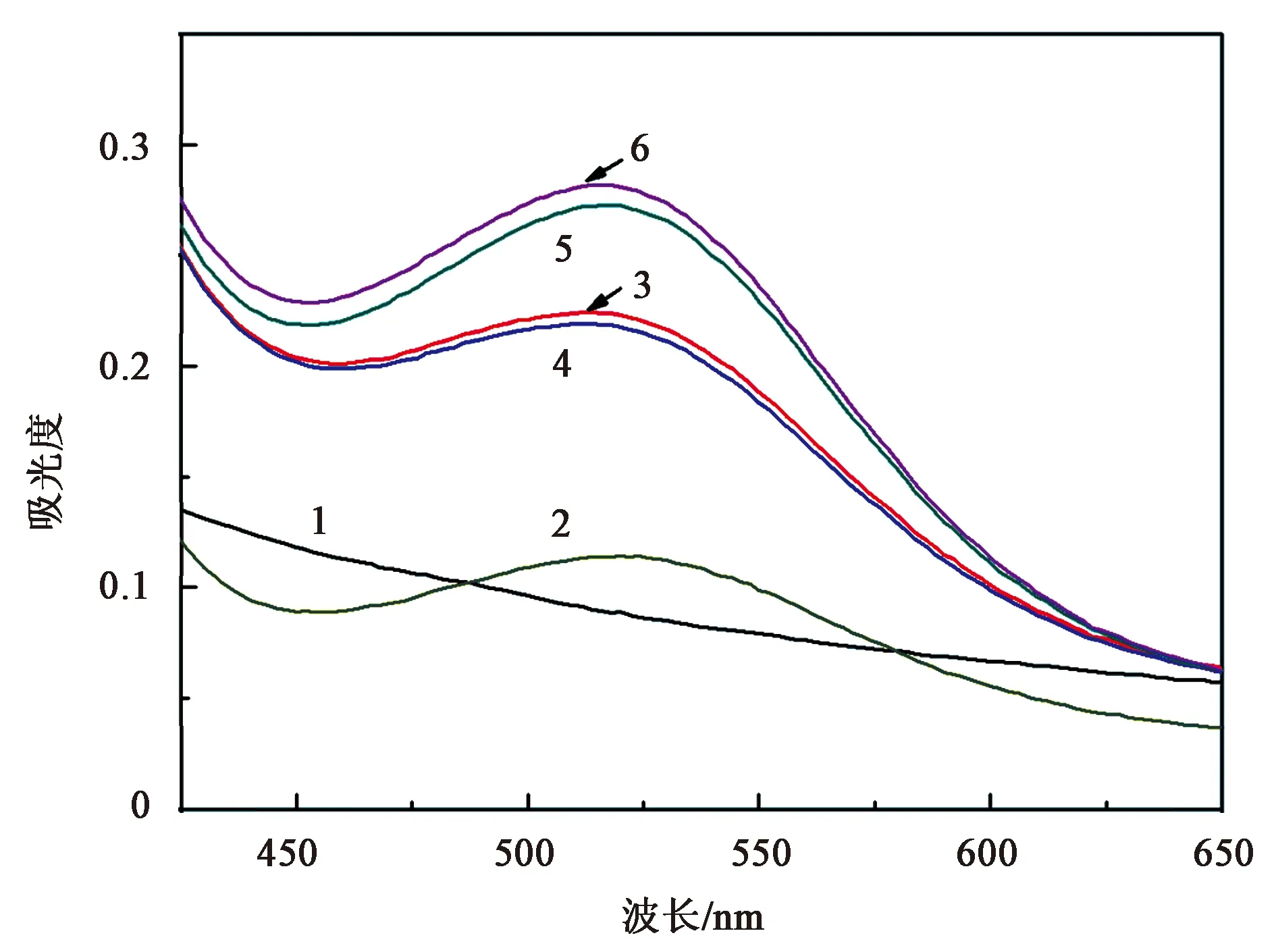
样1—Fe3O4溶液;样2—H2O2 + TMB;样3—Fe3O4 + H2O2 + TMB;样4—G4 + Fe3O4 + H2O2 + TMB;样5—Pb2+ + G4 + Fe3O4 + H2O2 + TMB;样6—Cd2+ + G4 + Fe3O4 + H2O2 + AAP。图2 基于Fe3O4过氧化物酶催化活性检测Cd2+和Pb2+重金属离子的可行性分析Fig.2 Feasibility verification of the developed colorimetric sensors for Cd2+ and Pb2+ detection based on the peroxidase activity of Fe3O4
2.3 实验条件优化
2.3.1缓冲溶液pH及浓度 Fe3O4模拟酶催化活性与反应介质密切相关,如图4A,pH较低时,随pH增加(3.0~3.5)检测信号增强;当pH升至3.5~4.0时检测信号达到最强;当pH继续增加时(4.0~5.0),检测信号不再增强反而减弱。其原因可能是,磁珠的催化活性及其与底物之间的反应与反应体系中的电子密度密切相关。因此,后续以pH 3.5的缓冲溶液进行实验。
缓冲液浓度对重金属离子检测的影响如图4B,缓冲液浓度为1~3.5 mmol/L时,随着缓冲液浓度增加传感信号增强;当缓冲液浓度达到3.5~8.0 mmol/L时,传感信号达到最大;若继续升高缓冲液浓度,检测信号则减弱。结果表明,缓冲液浓度为3.5~8.0 mmol/L的反应体系更适合于本法对靶标离子的检测。因此,选择3.5 mmol/L NaAc-HAc 进行后续实验。
2.3.2底物浓度 本文考察了不同浓度AAP和H2O2对检测信号的影响,如图5A所示。当底物AAP浓度很低时,检测信号较弱。随着底物浓度从1 mmol/L逐渐增加至50 mmol/L,检测信号变强。当再进一步增加底物浓度时,信号不再增强反而减小。
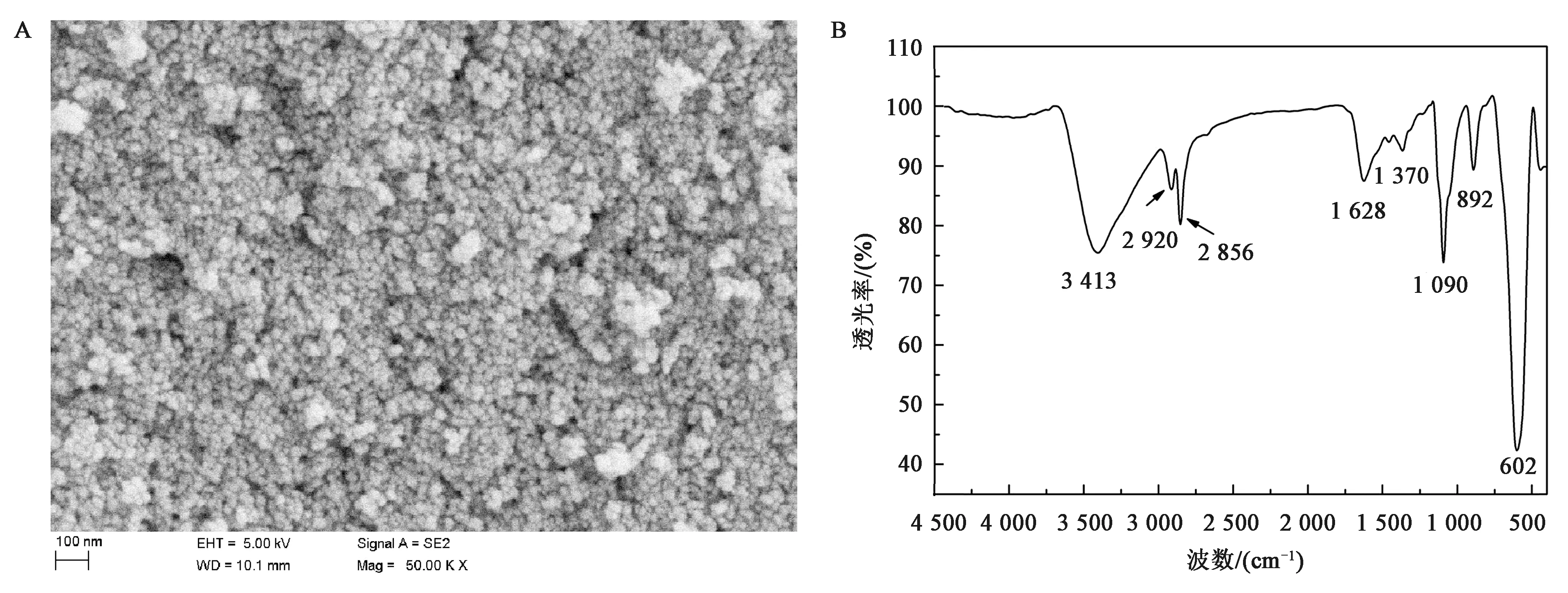
A:SEM电镜图;B:FT-IR能谱表征图图3 Fe3O4的SEM电镜和FT-IR能谱表征图Fig.3 SEM image and FT-IR spectrum of Fe3O4
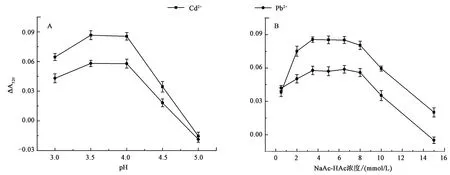
A:缓冲溶液pH的优化;B:NaAc-HAc浓度的优化图4 缓冲溶液pH及浓度的优化Fig.4 The optimization of pH and concentration of buffer solution
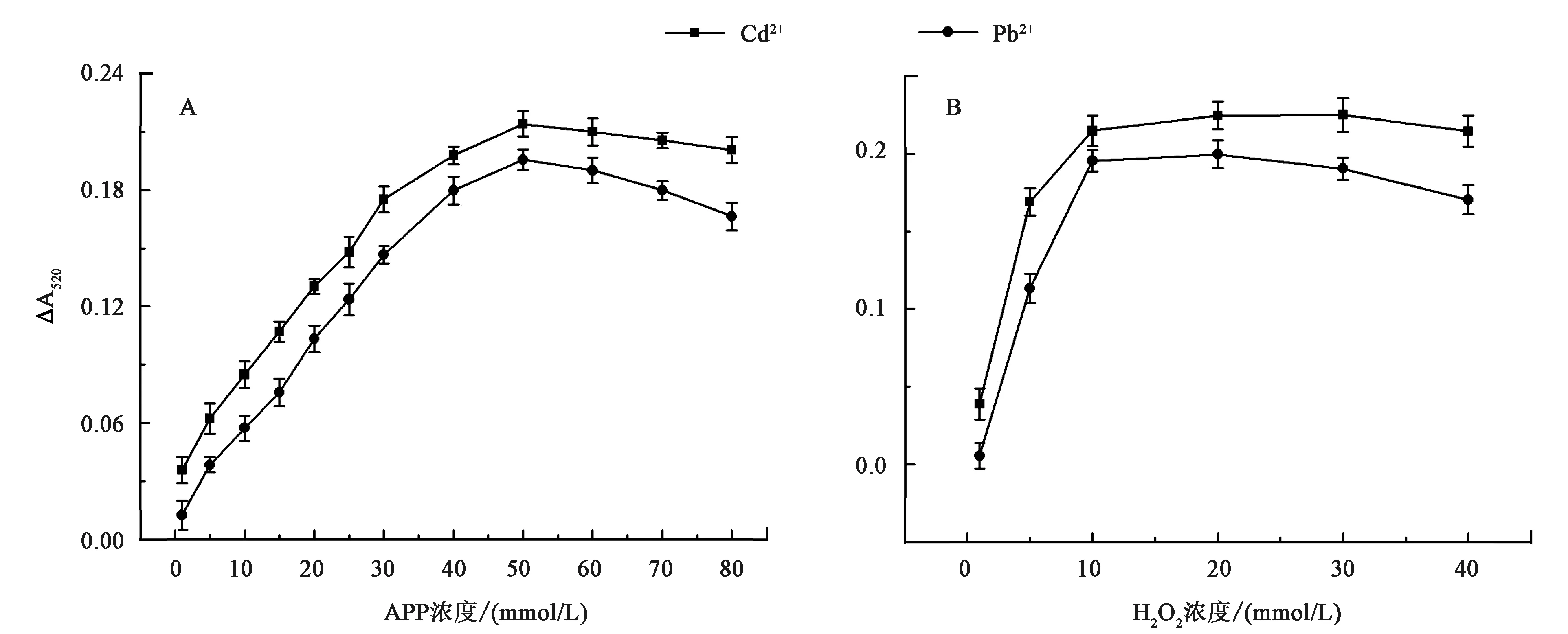
A:底物AAP浓度的优化;B:H2O2浓度的优化图5 反应体系中底物AAP和H2O2浓度的优化Fig.5 The optimization of AAP and H2O2 concentration in reaction system
对于H2O2,当其浓度为1 mmol/L时检测信号不明显(图5B)。随着H2O2浓度从1 mmol/L增加至10 mmol/L,检测信号显著增强并达到最大。而当H2O2浓度进一步增加时(10~40 mmol/L),检测信号基本稳定,不再随着H2O2浓度的增加而发生变化。说明H2O2可以促进纳米酶催化底物,直至酶与底物之间的反应达到饱和。因此,后续选择10 mmol/L H2O2进行实验。
2.3.3磁珠浓度 通过对反应体系中磁珠浓度进行优化,其结果如图6所示。在5~30 μg/mL Fe3O4浓度范围内,随磁珠浓度增加检测信号增强,且于30 μg/mL时Cd2+的检测信号达到最大值,而Pb2+的检测信号在30~60 μg/mL Fe3O4范围内出现最大值。之后,随Fe3O4浓度增加检测信号不再增强反而下降。可能的原因是,当纳米酶催化还未达到饱和时,增加酶的浓度有助于检测信号增强。而当酶与底物反应达到饱和之后,再增加酶的浓度可能会增加检测背景信号,造成检测信号降低。所以,以30 μg/mL Fe3O4作为最优浓度。
2.3.4核酸浓度 核酸适配体能与特定靶标发生特异性结合,因此在检测过程中使用适配体对检测特异性的增强非常重要。Liu等[36]曾对各种重金属离子适配体进行比较,发现G4与Cd2+和Pb2+的结合能力相对较强,所以本实验使用G4序列作为靶标识别元件。体系中核酸浓度的优化结果如图7所示,当G4浓度为10~20 nmol/L时,体系检测信号很强;但随着体系中核酸浓度的增加(从20~50 nmol/L),检测信号逐渐下降。下降的原因可能是,过多的核酸浓度紧紧包裹在磁珠表面,导致了其催化活性不易受靶标的调控。此外,随核酸浓度加大,Cd2+的检测信号比Pb2+的下降得更多。这可能是因为,一个Cd2+只能与一个G4结合,而一个Pb2+却能与多个G4结合,导致等量的靶标对G4的结合量不等,所以核酸浓度对体系信号的影响情况不一致。在后续实验中,以10 nmol/L的核酸作为最优浓度。
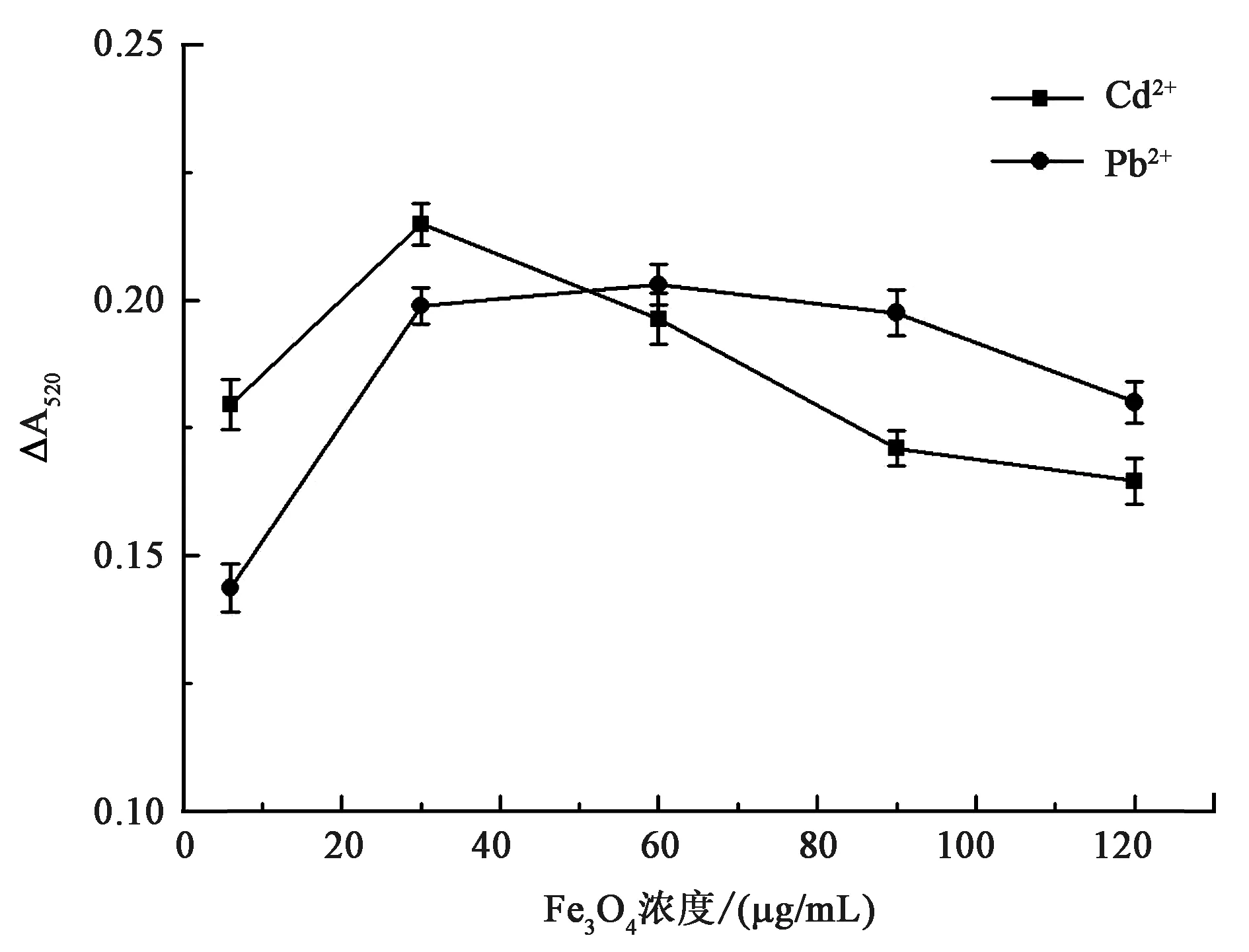
图6 反应体系中Fe3O4浓度的优化Fig.6 The optimization of Fe3O4 concentration in reaction system
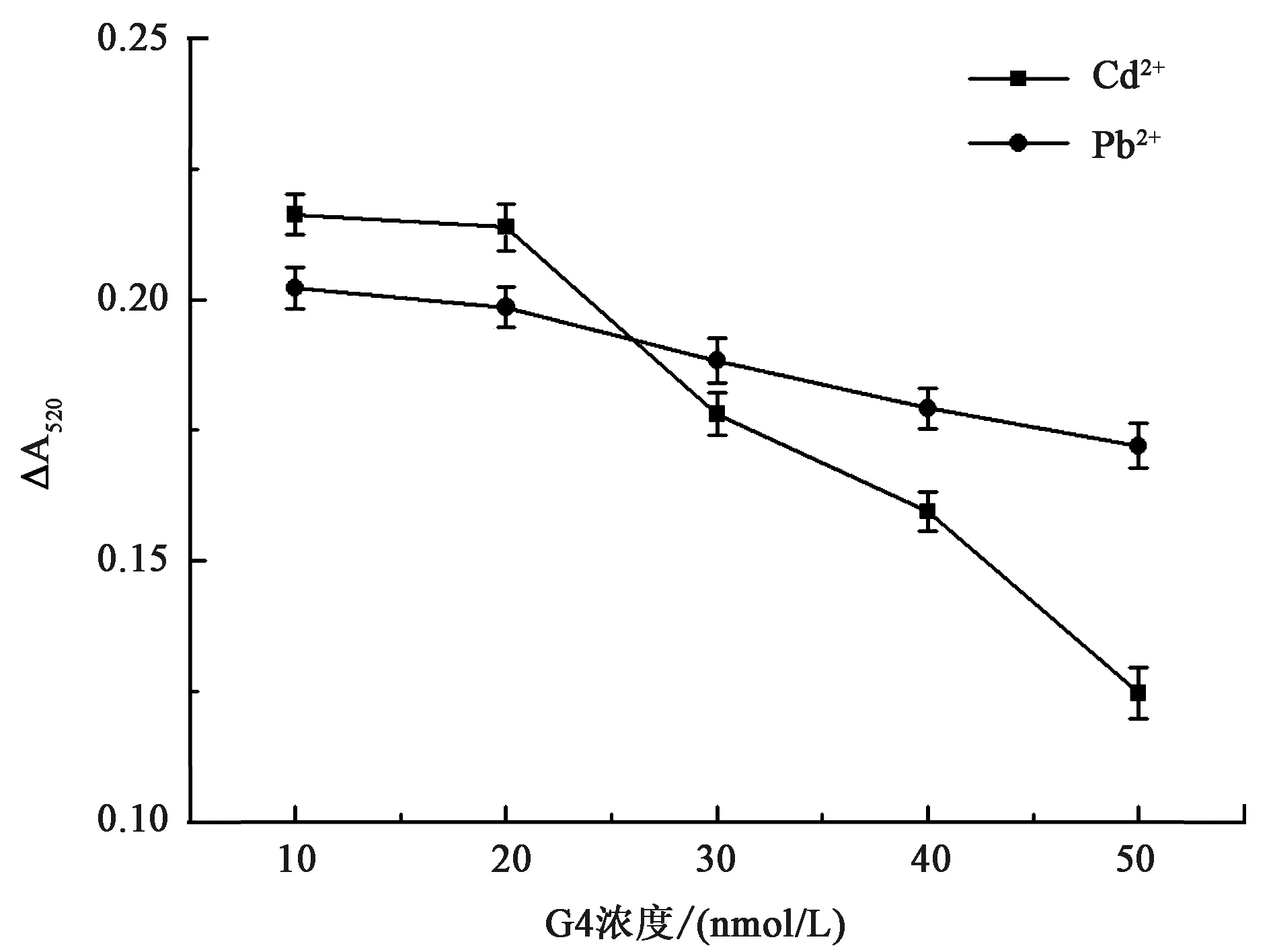
图7 反应体系中G4核酸浓度的优化Fig.7 The optimization of G4 concentration in reaction system
2.3.5孵育时间 如图8所示,在10~50 min内,检测信号随孵育时间的增加而逐渐增加直至50 min。当孵育时间超过50 min之后,体系检测信号达到平稳,不再随着孵育时间的增加而增加。因此,后续以50 min作为孵育时间。此外,Cd2+的检测信号与Pb2+的类似,但其检测信号始终比Pb2+强。
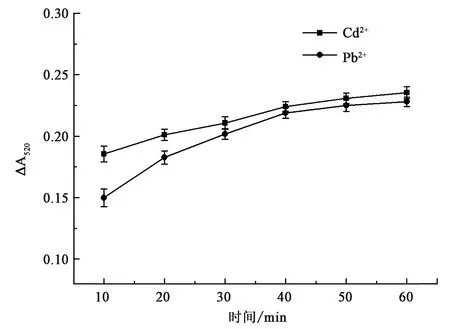
图8 孵育时间的优化Fig.8 The optimization of incubation time
2.4 灵敏度实验
分别往体系中加入不同浓度Cd2+(0.1~60 μmol/L)和Pb2+(0.1~60 μmol/L)离子,记录400~800 nm的吸收光谱,计算其520 nm处的吸光差值ΔA520。如图9A所示,随着Cd2+浓度增加,体系520 nm处吸收峰逐渐升高,检测信号ΔA520逐渐增强,当Cd2+浓度超过60 μmol/L后,其检测信号逐渐趋于平缓(图9C)。在低浓度(0.1~0.9 μmol/L)条件下,本方法检测信号ΔA520与体系中Cd2+浓度呈线性关系,线性方程为y=0.008+0.236x,其相关系数为0.9985。根据3σ/slope计算,得到本方法对Cd2+的最低检测限LOD=0.108 μmol/L。
同样的,按上述方法对Pb2+灵敏度进行测试。与Cd2+类似,随着溶液中Pb2+浓度增加,体系在520 nm处的吸收峰逐渐升高(图9B),其检测信号ΔA520也逐渐增强,当Pb2+浓度超过40 μmol/L后,其检测信号逐渐趋于平缓(图9D)。在低浓度(0.1~1 μmol/L)条件下,本方法检测信号ΔA520与体系中Pb2+浓度呈线性关系,线性方程为y=0.0092+0.2181x,其相关系数为0.9972。根据3σ/slope计算,得到本方法对Pb2+的最低检测限LOD=0.117 μmol/L。
2.5 特异性实验
为研究本传感体系对Cd2+和Pb2+重金属离子的检测特异性,分别向体系中加入等量(1 μmol/L)的竞争性离子(Zn2+、Fe2+、Hg2+、Ag+、As3+、Al3+、Cu2+和Cr3+)进行实验,测量其吸收光谱,计算检测信号ΔA520,结果如图10所示。含Cd2+和Pb2+的反应体系溶液为红色,而不含任何靶标的空白组和含其他竞争性离子的试验组体系溶液基本为浅黄色(图10)。因此,仅仅通过溶液颜色即可初步判断样品中Cd2+和Pb2+是否超标。此外,通过测定其520 nm处的吸光信号ΔA,发现含Cd2+和Pb2+的试验组其检测信号远远高于其他的竞争性靶标。因此,通过测定其520 nm处的吸光信号ΔA也可判断Cd2+和Pb2+离子是否超标。
3 讨论
本研究基于磁性Fe3O4纳米酶催化活性建立了一种Cd2+和Pb2+重金属离子比色检测法。当Cd2+和Pb2+存在时,体系溶液为红色;当没有Cd2+和Pb2+存在时,体系溶液为浅黄色或趋于无色。最优检测条件为:3.5 mmol/L NaAc-HAc缓冲溶液(pH 3.5)、50 mmol/L AAP、10 mmol/L H2O2、30 μg/mL Fe3O4、10 nmol/L G4和 50 min的孵育时间。当Cd2+和Pb2+浓度在0.1~0.9 μmol/L和0.1~1 μmol/L时,其最低检测限分别为0.108 μmol/L、0.117 μmol/L。本方法受竞争性离子(Zn2+、Fe2+、Hg2+、Ag+、As3+、Al3+、Cu2+和Cr3+)的干扰较小,特异性较强。
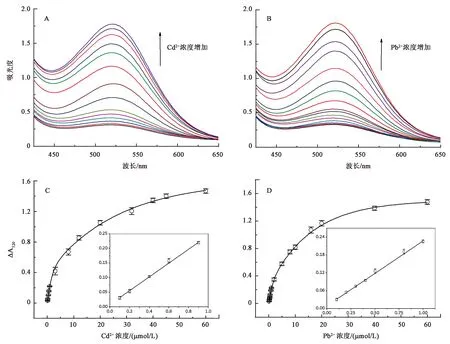
A,B:分别为传感器对不同Cd2+和Pb2+浓度的吸收光谱图;C,D:分别为对应的传感信号值曲线图(插图为线性关系图)。图9 基于磁性Fe3O4纳米酶催化活性的Cd2+和Pb2+重金属离子灵敏度检测Fig.9 Sensitivity of the present colorimetric sensors for Cd2+ and Pb2+ detection based on the peroxidase activity of Fe3O4
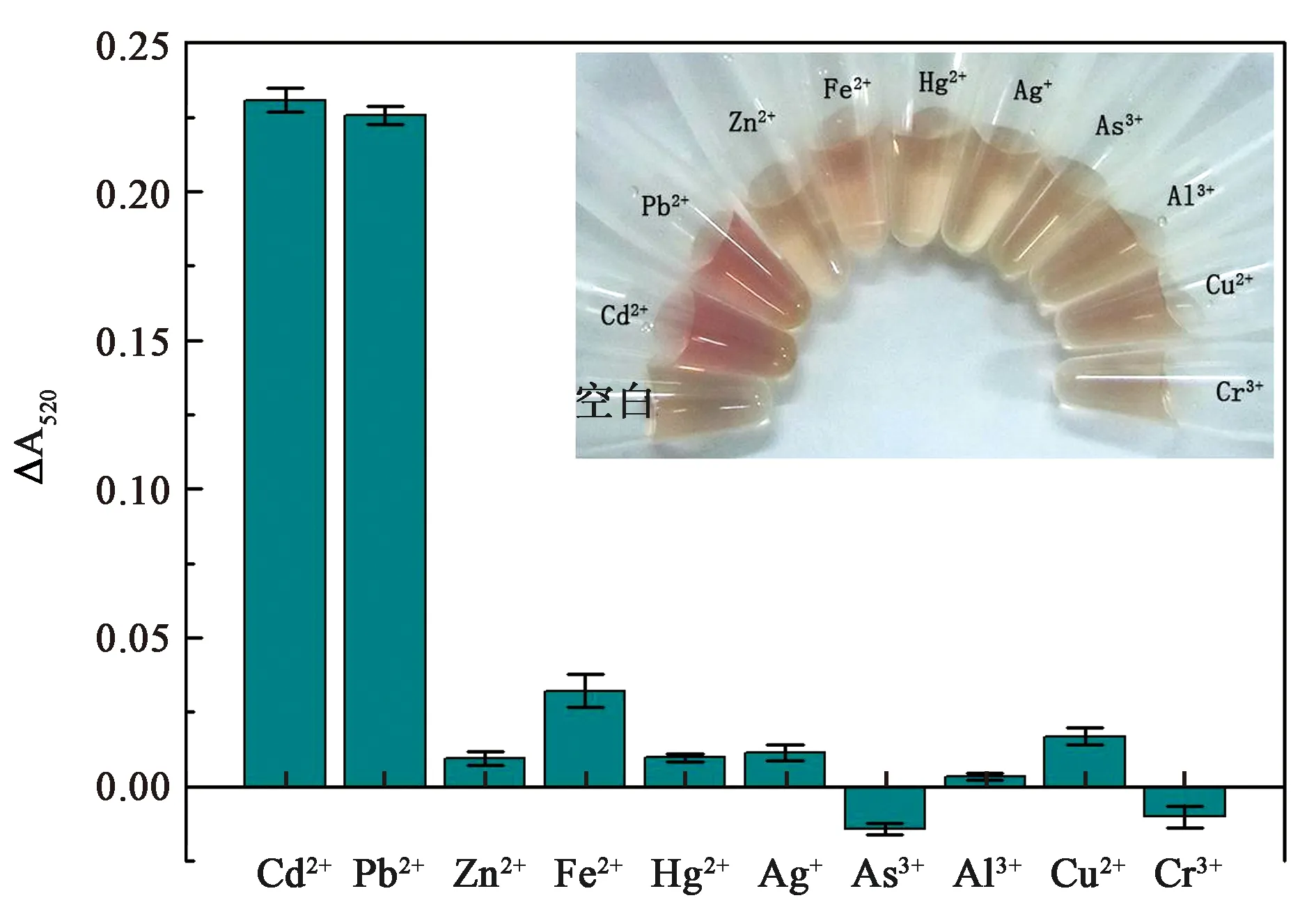
图10 基于磁性Fe3O4纳米酶催化活性的Cd2+和Pb2+重金属离子特异性检测Fig.10 Specificity of the present colorimetric sensors for Cd2+ and Pb2+ detection based on the peroxidase activity of Fe3O4
总之,本文建立的Cd2+和Pb2+重金属离子比色检测法具有一定的优点,如无需昂贵检测仪器和复杂操作过程、检测成本低等,因此更适合于发展中国家或偏远地区的实时在线监测。但本方法还存在一定的不足,虽核酸或适配体序列能一定程度提高特异性检测,但实际样品中往往存在着多种未知的阴阳离子和有机物,如何进一步提升本方法或该类方法的特异性依然有待研究。
