木质纤维素的微生物降解
2019-12-09许从峰艾士奇申贵男袁媛晏磊王伟东
许从峰,艾士奇,申贵男,袁媛,晏磊,王伟东
综 述
王伟东 黑龙江八一农垦大学教授,博士生导师,黑龙江省杰出青年基金获得者。黑龙江省“寒区环境微生物与农业废弃物资源化利用”重点实验室主任、黑龙江省“秸秆资源化利用工程技术研发中心”副主任、黑龙江省高校“寒区农业废弃物资源化利用科技创新团队”带头人。中国微生物学会环境微生物专业委员会委员,中国生态学会微生物生态专业委员会委员,中国植物营养与肥料学会生物与有机肥专业委员会委员,黑龙江省政府科顾委委员。研究方向为农业与环境微生物、农业废弃物资源化利用技术与工程。主持和承担国家自然科学基金等国家级课题6项,主持黑龙江省重大科技攻关项目、黑龙江省杰出青年科学基金等省部级课题6项;获省部级科学技术二等奖2项、三等奖3项;发表SCI收录论文20多篇,授权发明专利5项。

木质纤维素的微生物降解
许从峰,艾士奇,申贵男,袁媛,晏磊,王伟东
黑龙江八一农垦大学 生命科学技术学院 黑龙江省寒区环境微生物与农业废弃物资源化利用重点实验室,黑龙江 大庆 163319
木质纤维素广泛存在于自然界中,因结构复杂,其高效降解需要多种微生物的协同互作,由于参与木质纤维素降解的微生物种类繁多,其协同降解机理尚不完全明确。随着微生物分子生物学和组学技术的快速发展,将为微生物协同降解木质纤维素机制的研究提供新的方法和思路。笔者前期研究发现,细菌复合菌系在50 ℃下表现出强大的木质纤维素降解能力,菌系由可分离培养和暂时不可分离培养细菌组成,但是可分离培养细菌没有降解能力。通过宏基因组和宏转录组研究表明,与木质纤维素降解相关的某些基因表达量发生显著变化,通过组学方法有可能更加深入解释微生物协同降解木质纤维素的微生物学和酶学机理。文中从酶、纯培养菌株和复合菌群三个方面综述了木质纤维素微生物降解研究进展,着重介绍了组学技术在解析复合菌群作用机理方面的现状和应用前景,以期为探索微生物群落协同降解木质纤维素的机理提供借鉴。
木质纤维素,复合菌群,协同作用,降解机理
木质纤维素是世界上产量最大的可再生生物质资源,全世界每年产量达到1 500亿t,仅秸秆产量就达到60亿t,中国秸秆年产量11.13亿t,它的有效处理和资源化利用对于社会可持续发展意义重大[1]。木质纤维素的生物法利用一直是其主要的资源化利用方式,前提是获取高效降解木质纤维素的微生物,明确微生物个体和群体之间协同作用规律。木质纤维素结构异常复杂,其降解需要多种微生物的协同互作,很多微生物分类和代谢类型多样性等还不明确,以及微生物间协同降解机制尚未解析,极大地阻碍木质纤维素降解微生物资源的开发与应用[2]。
从早期的真菌纤维素酶开始,研究人员为揭示木质纤维素的微生物降解机理一直在不断努力[3-4],在真菌和细菌的纯培养菌株、木质纤维素降解酶、纤维素小体 (Cellulosome) 和微生物复合菌群协同作用等方面的研究取得较大的进展。随着组学技术的发展,多维组学分析可准确预测降解木质纤维素菌群内部的共生或协同关系,极大地扩展对微生物群落的认知,采用传统微生物学与多维组学研究方法结合,有望取得更大的突破。
1 降解木质纤维素的酶和微生物
1.1 降解木质纤维素的酶
自然界中存在极其复杂的微生物群落,能够产生数量庞大、种类丰富的木质纤维素降解酶,它们结构各异,作用方式不同,但是根本原理均在于酶破坏化学键致使木质纤维素结构裂解[5-6]。
木质纤维素主要由纤维素、半纤维素和木质素构成,各成分差异较大,对应降解酶也不尽相同。纤维素酶包括内切葡聚糖酶、外切葡聚糖酶和β-葡萄糖苷酶等,其中内切酶作用于纤维素的无定形区,产生纤维素反应末端,外切酶作用于纤维素的结晶区,产生纤维二糖或葡萄糖,β-葡萄糖苷酶将纤维寡糖水解为葡萄糖是被普遍接受的纤维素降解理论 (图1),研究表明部分厌氧菌产生的纤维素酶通过锚定结构域与支架蛋白上黏附结构域特异性结合,组装成多酶聚合体——纤维素小体,能够有效缩短细胞与底物的距离,实现酶循环利用和产物直接矿化[8-9]。
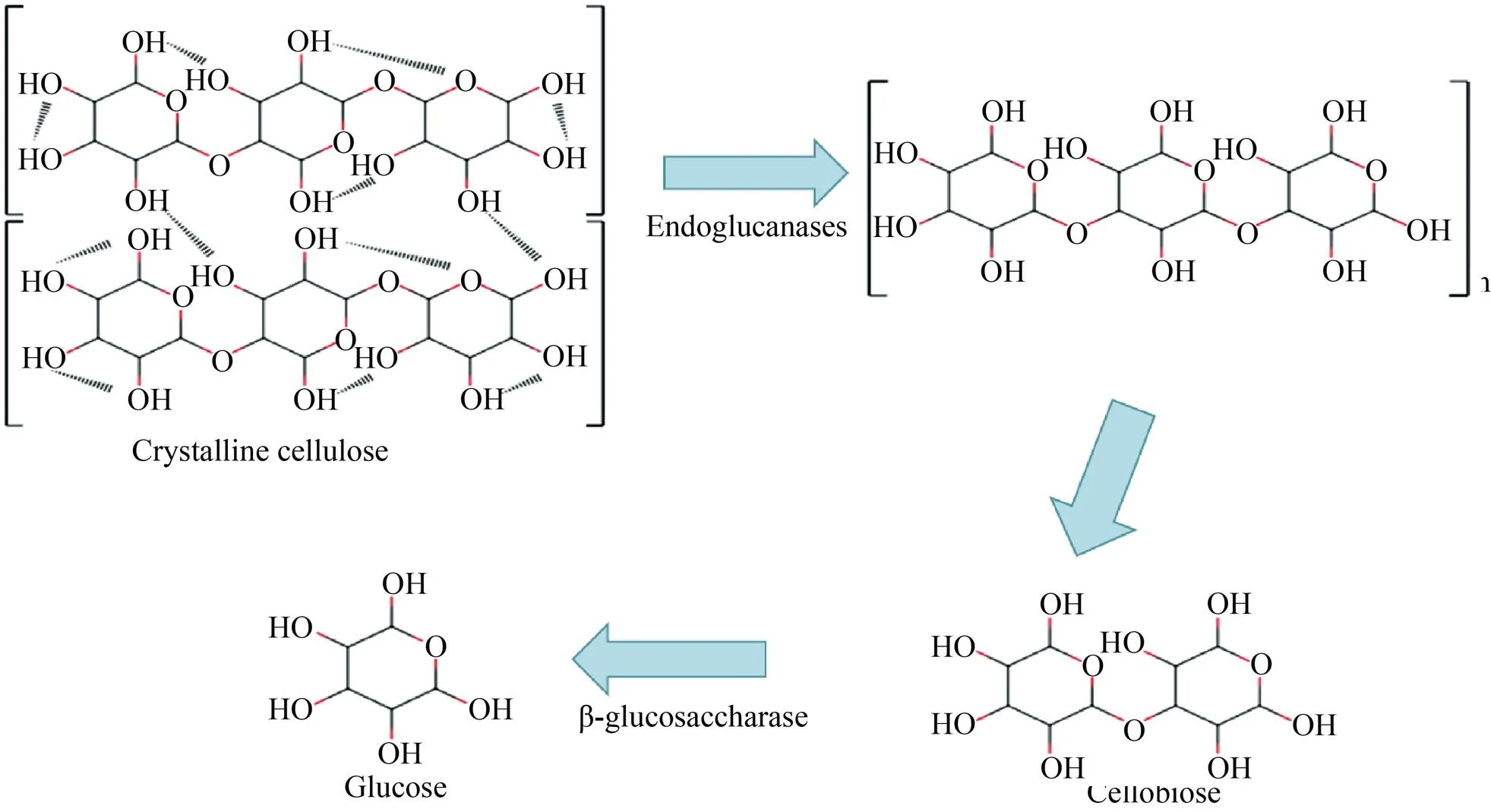
图1 纤维素降解机理[7]
木聚糖是半纤维素主要成分,是一类复杂多聚五碳糖,其降解酶主要有内切β-木聚糖酶 (作用于木聚糖主链)、木聚糖外切酶[10](作用于寡聚木糖和木聚糖的非还原端) 和辅酶[11](作用于木聚糖的支链)。木质素是含有氧代苯丙醇及其衍生物结构单元的、无定形的、具有芳香性的高聚物,很难被微生物降解。木质素降解酶主要有漆酶、锰过氧化物酶、木质素过氧化物酶及辅酶,木质素在微生物解聚过程中形成许多丁香酚基单元、愈创木基单元和p-羟苯基单元,同时也破坏各种连接键,从而将木质素降解为各种小分子片段,最后进入三羧酸循环 (图2)。木质纤维素降解酶的种类很多,包括各种高温、高盐、强碱和高压等极端环境降解酶,但单种酶降解能力有限,需要多种酶共同作用。
1.2 降解木质纤维素纯培养菌株
环境中存在丰富的降解木质纤维素微生物,主要包括细菌和真菌,它们种类繁多,相互协调,其中大部分难以培养,已分离200多种,主要来源于堆肥、瘤胃、厌氧污泥和土壤的微生物菌群[13]。细菌具有繁殖周期短、结构简单、抗逆性强和耐酸碱等优点,在降解木质纤维素方面具有巨大应用潜力[14],根据对氧气依赖程度可分为好氧菌与厌氧菌,好氧菌如噬纤维菌[15]、假单胞菌[16]、热酸菌[17]和芽孢杆菌[18]等,厌氧菌有梭菌[19]、热解纤维素菌[20]和醋弧菌[21]等。大部分好氧细菌分泌纤维素胞外酶,种类单一,降解效率低,实用性弱[22]。厌氧菌具有降解效率高、不易被杂菌污染等优点,但是生长速度缓慢,降解中间产物如戊糖、甲酸等对其有抑制作用[23]。
自然环境中真菌是降解木质纤维素的主要微生物,已报道的真菌包括曲霉[24]、木霉[25]和瘤胃真菌[26]等,由于其生产木质纤维素酶的效率低、酶活不高和致病性 (曲霉) 等缺点,限制了真菌的大规模开发和使用。另外,繁殖缓慢、降解能力弱的放线菌已经被发现,如小单胞菌[27]和诺卡氏菌[28]等。随着基因工程的发展,菌株稳定性和整体酶系的协同性得到一定的加强,将工程菌以不同比例混合,有望应用到实际生产中[29]。
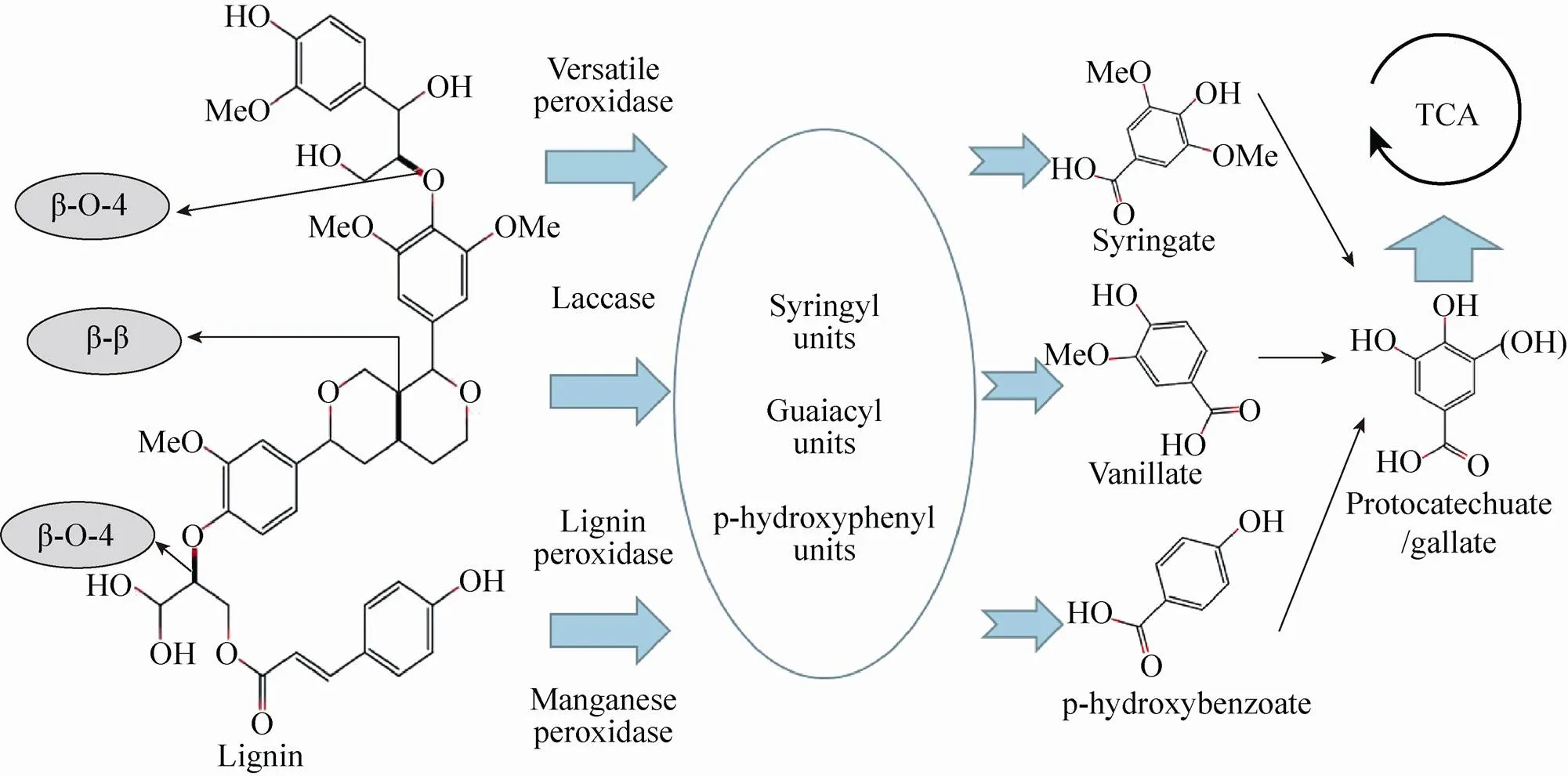
图2 木质素降解机理[12]
1.3 降解木质纤维素复合菌群
复合菌群是由多种微生物组成的群落,主要来源于自然界筛选和菌株组配,利用复合菌群解决木质纤维素降解难的问题是科研工作者密切关注的重点[30-31]。复合菌群形成动态、多变的微生物环境,具有极其丰富多样的生态适应能力和生理代谢功能,木质纤维素降解正需要依靠复合菌群内部的协同互作。
1.3.1 降解木质纤维素纯培养菌株组合菌群
组合菌群的研究在菌种的筛选和菌群的组装效果方面报道较多,为了更好地研究菌群降解木质纤维素的协同机制,组合群落需要依据实验目的进行严格设计,考虑菌群中物种组成和比例、酶的种类、代谢产物以及基因组信息等因素[32-34]。Zhang等[35]将枯草芽孢杆菌(AY881635)和煎盘梭菌(NR026490) 组合,获得一组兼性厌氧高效降解木质纤维素菌群M1,降解能力远高于纯培养菌株;Puentes-Téllez等[36]采用基于分子表型、鉴定和代谢特性的方法从甘蔗渣中筛选出18株木质纤维素降解菌,结合生态学理论和富集原理,组配出高效降解木质纤维素菌群MAMC,根据MAMC的降解能力和功能多样性测定结果,发现随着物种组成多样性的增加,木质纤维素的降解率显著上升。
获得降解木质纤维素菌株是组合菌群的基础,保证组合菌群能够高效降解木质纤维素是关键。在实际操作中,尽管有许多分析的手段,但要构建一个理想的组合菌群还是要尽可能从不同生境分离和培养降解能力强的微生物[37]。微流控技术有效降低共生以及互生菌筛选难度,为功能菌株的获取提供新的途径[38],同时,最好对获得可培养新型菌株进行基因组测序,除了考虑单菌基因组,组合菌群基因组信息对于研究不同菌株与木质纤维素降解的联系也十分必要[39]。构建降解木质纤维素菌群时有以下问题需要注意:(1) 菌株生长周期长短;(2) 菌株之间是否相互拮抗;(3) 菌株混合比例是否合适;(4) 降解效率是否在传代过程中衰退。
1.3.2 降解木质纤维素天然复合菌群
从环境中筛选高效降解木质纤维素的微生物菌群,通过定向优化进一步加强木质纤维素降解效率,研究其物种组成及降解功能,获取木质纤维素降解相关基因信息,将为研究菌群降解木质纤维素协同机理提供新的思路和方案。崔宗均等[40]采用酸碱互补原则选育出复合菌群MC1 3 d内可降解98%的脱脂棉和94%的滤纸;王伟东等[41]通过限制性培养技术从堆肥中获得了一组50 ℃静置条件下3 d可完全降解滤纸的复合菌群WSC-9;Liang等[42]从菌糠中获得复合菌群OEM2 12 d内可降解41.5%的水稻秸秆和85%的半纤维素;Lu 等[43]从厌氧消化污泥中获得一组高温降解木质纤维素菌群TC-Y 20 d内降解49.5%的玉米秸秆,其中纤维素、半纤维素和木质素的降解率分别为52.76%、62.45%和42.23%。
降解木质纤维素复合菌群中微生物之间具有协同关系,其木质纤维素降解能力、抗逆性及稳定性远远超过纯培养菌株和组合菌群。前期的木质纤维素降解理论主要以真菌的研究为基础,随后得到不断的补充和改善,但是木质纤维素降解菌群关系复杂多变,协同作用研究进展缓慢[44],主要有以下几个原因:1) 物种组成不稳定,很难控制;2) 群落组成未明确,协同关系不清晰;3) 菌群功能多样性,代谢途径错综复杂;4) 研究理论匮乏,技术手段待完善。
2 组学方法揭示木质纤维素降解机理
复杂微生物群落降解木质纤维素机制的揭示具有挑战性,利用先进的分析技术,如宏基因组学[45]和宏转录组学[46]等,将为解析复杂微生物群落降解功能和代谢途径提供新的切入点[47-48]。
2.1 宏基因组学
宏基因组学研究方法最早应用于基因序列的分析和功能筛选[49]。近年来,随着测序技术和分析工具的迅猛发展,采用新一代测序技术研究降解木质纤维素菌群基因,能快速准确获得海量微生物基因数据以及更高的分类学信息,是研究微生物菌群功能的重要途径[50-51]。
应用于木质纤维素降解菌群的研究可发现新型降解基因以及代谢通路的调控机制。Dai等[52]利用宏基因组测序与基于人工染色体功能筛选方法相结合发现牦牛瘤胃中有150种降解木质纤维素的糖苷水解酶基因,大多数与编码相关功能的基因聚类较近,其中25个家族来自拟杆菌门、 4个来自厚壁菌门;Thornbury等[53]从北美豪猪体内利用宏基因组技术通过与纤维素和半纤维素降解相关的保守催化域识别出4种新型木质纤维素降解酶基因,分别是β-葡糖苷酶、β-L-阿拉伯呋喃糖酶、β-木糖苷酶和内切4-β-木聚糖酶,并成功在大肠杆菌中表达;Wilhelm等[54]利用宏基因组技术结合同位素追踪改良基因组装方法,发现7 500多个包含独特的碳水化合物活性基因簇。
宏基因组测序可以直接获得微生物功能基因的相对丰度,鉴定分辨率可达种的水平,通过基因分箱技术获得其中降解木质纤维素关键菌株较完整的基因组,形成假定的功能通路和模块,从而挖掘微生物功能和遗传变异性[55-57]。但是,宏基因组测序技术具有自身局限性,如宏基因组学以DNA为研究对象,并不能区分其来源于有生命或无生命的生物体,受组装方式影响,得到的微生物菌群基因组信息往往不够准确[58]。
2.2 宏转录组学
宏转录组学主要研究微生物群落基因转录情况及调控规律,功能基因分析可以说明其是否已经表达以及表达量的多少,有效说明菌群活力大小,是分析微生物群落代谢能力的重要手段。宏转录组是以菌群全部RNA为研究对象,仅鉴别活体生物,排除休眠或死亡微生物对结果的影响,不仅可以捕捉菌群的动态变化,还可以估算群落中哪些微生物正在进行高效转录[59]。
国内外已存在利用宏转录组高通量测序技术研究木质纤维素降解的相关报道[60-61]。Gruninger 等[62]利用宏转录组技术研究、、和对碳水化合物的消化,发现所有物种均表达大量编码碳水化合物活性酶的转录本,占转录组的8.3%–11.3%,其中参与半纤维素消化的酶家族数量最多;Janusz 等[63]对生长在桦木、白蜡树、枫树木屑和液体培养基 (对照) 上的真菌的转录组进行了分析,发现液体培养基中检测到真菌的特殊转录本数量最高 (107),而在含有枫树木屑的真菌培养中,检测到特殊转录本数量最低(11),白蜡树木屑培养基上生长的真菌中,上调转录本数量最多(828),在294个可能参与木质纤维素降解的基因中,59个基因表达发生了显著变化 (<0.01);Peng等[64]比较分析了22个与木质纤维素降解相关的担子菌转录组数据集,鉴定了328个常见的诱导基因和318个抑制基因,并定义了一组核心的碳水化合物活性酶,该酶被大多数担子菌共享。
研究特定环境下基因的表达谱,能将微生物菌群与其功能联系到一起,从而更好地了解微生物群落的代谢活性,但是宏转录组并不完美,比如费用较高,样品制备和分析过程复杂,极大地阻碍研究速度。微生物基因转录率的高低会让数据结果有偏差,需要与菌群DNA数据相结合,才能获得准确的微生物丰度变化和转录情况[65]。另外,RNA不仅含量极低,还容易降解和污染,在指定环境中提取总RNA时要格外小心[66-67]。
2.3 多维组学
微生物群落协同关系的变化跟菌群内部化学物质合成速率、基因转录表达以及蛋白质活性均息息相关[68]。多维组学以菌群为核心,利用宏基因组、宏转录组、蛋白质组和代谢组等技术联合空间相关性分析、稀疏典型相关分析、相关网络分析、代谢活性分析、普氏分析和多重共惯性分析等方法,从多角度、多层次剖析微生物菌群功能,给降解木质纤维素菌群研究提供更完整、更系统的生物学分析方法,是非常有前景的研究方向 (图3)。但是,整合多组学数据困难重重,例如微生物降解木质纤维素过程中基因表达与代谢物来自不同的降解时期[70],而且代谢底物可能是不同菌种之间协同作用的结果[71];宏基因组和代谢组基因分类信息联系紧密[72-74];多维组学数据的分析工具和方法尚不完善[75]。
分析降解木质纤维素菌群组学数据只是开始,建立菌群代谢物与组学数据之间的联系是下一阶段的主要目标。微生物代谢物可影响其他共生菌,从而决定整个菌群的功能,然而,鉴定微生物菌群中代谢物来源和收集瞬间代谢产物是非常困难的。在多组学分析中,数据集可能包括数量庞大的微生物和代谢物数据,分析过程中可能会出现偶然的相关,因此多重比较校正是关键,校正显著性检验的方法有假阳性率校正和总体错误率校正等[76]。
尽管存在诸多挑战,也有一些成功整合多维组学数据的例子,这些研究成果远超单组学分析。Alessi等[77]以降解麦秆微生物菌群为研究对象,结合宏转录组学与以质谱为基础的蛋白质组学,鉴定了1 127种蛋白质,揭示了广泛的水解纤维素酶、半纤维素酶和参与木质纤维素降解的碳水化合物结合模块;Hassa等[78]利用多维组学对厌氧发酵中木质纤维素降解菌群进行研究,组装了数百个新菌的全基因组,揭示微生物群落遗传潜力,宏转录组数据为了解群落代谢活性提供视角,蛋白质组数据揭示木质纤维素降解群落成员表达的酶谱,鉴定纤维素和半纤维素降解酶以及其他聚合物的利用。综上所述,多维组学数据可以更全面地解析降解木质纤维素菌群基因组数据、木质纤维素降解酶和代谢物之间的关联,使研究结果更具有指导意义。

图3 利用多维组学分析菌群降解木质纤维素的过程[69](A:空间相关性分析;B:稀疏典型相关分析;C:相关网络分析;D:代谢活性网络分析;E:普氏分析;F:多重共惯性分析)
3 展望
复合菌群协同作用在木质纤维素降解方面具有优势已经成为共识,尤其是在开放环境中。从自然界富集菌群中定向筛选特定功能的纯培养菌株,根据他们各自的特点结合目标功能,有目的地进行菌群重构,虽然组配菌群木质纤维素降解能力比天然菌群有所下降,但是组配群体组成明确,便于分析研究。随着组学技术的发展,多组学分析将广泛应用于组配菌群,组配菌群的简易性加上多维组学分析的全面性,菌与菌之间、菌与环境之间的协同机制将会更加清晰。
深入理解组配菌群降解机制后,就可以人工调控菌群功能,更好地让组配菌群应用于实际生产。组配菌群在秸秆能源化、肥料化、饲料化、原料化和基质化,以及其他农林有机废弃物的资源化利用方面起到重要的作用,实现木质纤维素基础研究与应用研究的完美结合。
[1] 中华人民共和国国家统计局[EB/OL]. [2019-07-13]. http://data.stats.gov.cn/easyquery.htm?cn=C01&zb=A0D0F&sj=2018.
[2] Großkopf T, Soyer OS. Synthetic microbial communities. Curr Opin Microbiol, 2014, 18: 72–77.
[3] van Dijk EL, Auger H, Jaszczyszyn Y, et al. Ten years of next-generation sequencing technology. Trends Genet, 2014, 30(9): 418–426.
[4] Callahan BJ, Mcmurdie PJ, Holmes SP. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J, 2017, 11(12): 2639–2643.
[5] Wang Q, Awasthi MK, Zhao J, et al. Improvement of pig manure compost lignocellulose degradation, organic matter humification and compost quality with medical stone. Bioresour Technol, 2017, 243: 771–777.
[6] Cragg SM, Beckham GT, Bruce NC, et al. Lignocellulose degradation mechanisms across the tree of life. Curr Opin Chem Biol, 2015, 29: 108–119.
[7] Lynd LR, Weimer PJ, van Zyl WH, et al. Microbial cellulose utilization: fundamentals and biotechnology. Microbiol Mol Biol Rev, 2002, 66(3): 506–577.
[8] Artzi L, Bayer EA, Moraïs S. Cellulosomes: bacterial nanomachines for dismantling plant polysaccharides. Nat Rev Microbiol, 2016, 15(2): 83–95.
[9] Smith SP, Bayer EA, Czjzek M. Continually emerging mechanistic complexity of the multi-enzyme cellulosome complex. Curr Opin Struct Biol, 2017, 44: 151–160.
[10] Ahmed AAQ, Babalola OO, Mckay T. Cellulase- and xylanase-producing bacterial isolates with the ability to saccharify wheat straw and their potential use in the production of pharmaceuticals and chemicals from lignocellulosic materials. Waste Biomass Valoriz, 2017, 9(5): 765–775.
[11] Collins T, Gerday C, Feller G. Xylanases, xylanase families and extremophilic xylanases. FEMS Microbiol Rev, 2005, 29(1): 3–23.
[12] Boerjan W, Ralph J, Baucher M. Lignin biosynthesis. Ann Rev Plant Biol, 2003, 54(1): 519–546.
[13] Tsapekos P, Kougias PG, Vasileiou SA, et al. Bioaugmentation with hydrolytic microbes to improve the anaerobic biodegradability of lignocellulosic agricultural residues. Bioresour Technol, 2017, 234: 350–359.
[14] Hasunuma T, Okazaki F, Okai N, et al. A review of enzymes and microbes for lignocellulosic biorefinery and the possibility of their application to consolidated bioprocessing technology. Bioresour Technol, 2013, 135: 513–522.
[15] Zhu YT, Han LL, Hefferon KL, et al. Periplasmichutchinsonii endoglucanases are required for use of crystalline cellulose as the sole source of carbon and energy. Appl Environ Microbiol, 2016, 82(15): 4835–4845.
[16] Xu ZX, Qin L, Cai MF, et al. Biodegradation of kraft lignin by newly isolated,, andstrains. Environ Sci Pollut Res, 2018, 25(14): 14171–14181.
[17] Shahid S, Tajwar R, Akhtar MW. A novel trifunctional, family GH10 enzyme from11B, exhibiting endo-xylanase, arabinofuranosidase and acetyl xylan esterase activities. Extremophiles, 2018, 22(1): 109–119.
[18] Rastogi G, Bhalla A, Adhikari A, et al. Characterization of thermostable cellulases produced byandstrains. Bioresour Technol, 2010, 101(22): 8798–8806.
[19] Kothari N, Holwerda EK, Cai CM, et al. Biomass augmentation through thermochemical pretreatments greatly enhances digestion of switchgrass by. Biotechnol Biofuels, 2018, 11: 219.
[20] Brunecky R, Donohoe BS, Yarbrough JM, et al. The multi domainCelA cellulase excels at the hydrolysis of crystalline cellulose. Sci Rep, 2017, 7: 9622.
[21] Khan AW, Murray WD. Influence ofon cellulose degradation by. J Appl Microbiol, 2010, 53(3): 379–383.
[22] Jørgensen H, Pinelo M. Enzyme recycling in lignocellulosic biorefineries. Biofuel Bioprod Bior, 2016, 11(1): 150–167.
[23] Qing Q, Yang B, Wyman C E. Xylooligomers are strong inhibitors of cellulose hydrolysis by enzymes. Bioresour Technol, 2010, 101(24): 9624–9630.
[24] Ahmad R, Khare SK. Immobilization ofCellulase on multiwall carbon nanotubes for cellulose hydrolysis. Bioresour Technol, 2018, 252: 72–75.
[25] Du J, Zhang X, Li XZ, et al. The cellulose binding region incellobiohydrolase I has a higher capacity in improving crystalline cellulose degradation than that of. Bioresour Technol, 2018, 266: 19–25.
[26] Dollhofer V, Dandikas V, Dorn-In S, et al. Accelerated biogas production from lignocellulosic biomass after pre-treatment with. Bioresour Technol, 2018, 264: 219–227.
[27] Lee HJ, Whang KS.sp. nov., isolated from forest soil. Int J Systemat Evolut Microbiol, 2017, 67(6): 1746–1751.
[28] Bučko M, Vikartovská A, Gemeiner P, et al.cells immobilized in sodium alginate-cellulose sulfate-poly (methylene--guanidine) capsules: mechanical resistance and operational stability. J Chem Technol Biotechnol Biotechnol, 2010, 81(4): 500–504.
[29] Zhang HF. Construction of cellulase recombinant strains andsaccharification and fermentation of rice straw for ethanol production[D]. Hefei: Hefei University of Technology, 2017 (in Chinese).
张海峰. 纤维素酶基因工程菌构建及水稻秸秆原位糖化发酵产乙醇[D]. 合肥: 合肥工业大学, 2017.
[30] Yuan XF, Ma L, Wen BT, et al. Enhancing anaerobic digestion of cotton stalk by pretreatment with a microbial consortium (MC1). Bioresour Technol, 2016, 207: 293–301.
[31] Carpio IEM, Franco DC, Sato MIZ, et al. Biostimulation of metal-resistant microbial consortium to remove zinc from contaminated environments. Sci Total Environ, 2016, 550: 670–675.
[32] Busby PE, Soman C, Wagner MR, et al. Research priorities for harnessing plant microbiomes in sustainable agriculture. PLoS Biol, 2017, 15(3): e2001793.
[33] Chang C, Bowman JL, Meyerowitz EM. Field guide to plant model systems. Cell, 2016, 167(2): 325–339.
[34] Jiménez DJ, Dini-Andreote F, DeAngelis KM, et al. Ecological insights into the dynamics of plant biomass-degrading microbial consortia. Trends Microbiol, 2017, 25(10): 788–796.
[35] Zhang DD, Wang Y, Zheng D, et al. New combination of xylanolytic bacteria isolated from the lignocellulose degradation microbial consortium XDC-2 with enhanced xylanase activity. Bioresour Technol, 2016, 221: 686–690.
[36] Puentes-Téllez PE, Salles JF. Construction of effective minimal active microbial consortia for lignocellulose degradation. Microb Ecol, 2018, 76(2): 419–429.
[37] Bai Y, Müller DB, Srinivas G, et al. Functional overlap of theleaf and root microbiota. Nature, 2015, 528(7582): 364–369.
[38] Park J, Kerner A, Burns MA, et al. Microdroplet-enabled highly parallel Co-cultivation of microbial communities. PLoS ONE, 2011, 6(2): e17019.
[39] Müller DB, Schubert OT, Röst H, et al. Systems-level proteomics of two ubiquitous leaf commensals reveals complementary adaptive traits for phyllosphere colonization. Mol Cell Proteom, 2016, 15(10): 3256–3269.
[40] Cui ZJ, Li MD, Piao Z, et al. Selection of a composite microbial system MC1 with efficient and stability cellulose degradation bacteria and its function. Chin J Environ Sci, 2002, 23(3): 36–39 (in Chinese).
崔宗均, 李美丹, 朴哲, 等. 一组高效稳定纤维素分解菌复合系MC1的筛选及功能. 环境科学, 2002, 23(3): 36–39.
[41] Wang WD, Cui ZJ, Yang HY, et al. Stability of a composite microbial system WSC-6 with efficient cellulose degrading. China Environ Sci, 2005, 25(5): 567–571 (in Chinese).
王伟东, 崔宗均, 杨洪岩, 等. 高效稳定纤维素分解菌复合系WSC-6的稳定性. 中国环境科学, 2005, 25(5): 567–571.
[42] Liang JJ, Fang XX, Lin YQ, et al. A new screened microbial consortium OEM2 for lignocellulosic biomass deconstruction and chlorophenols detoxification. J Hazard Mater, 2018, 347: 341–348.
[43] Lu J, Yang ZM, Xu WY, et al. Enrichment of thermophilic and mesophilic microbial consortia for efficient degradation of corn stalk. J Environ Sci, 2019, 78(4): 118–126.
[44] Ai SQ, Zhao YQ, Sun ZY, et al. Change of bacterial community structure during cellulose degradation by the microbial consortium. Chin J Biotech, 2018, 34(11): 1794–1808 (in Chinese). 艾士奇, 赵一全, 孙志远, 等. 复合菌系降解纤维素过程中微生物群落结构的变化. 生物工程学报, 2018, 34(11): 1794–1808.
[45] Qin JJ, Lin YR, Cai ZM, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature, 2012, 490(7418): 55–60.
[46] Garza DR, Dutilh BE. From cultured to uncultured genome sequences: metagenomics and modeling microbial ecosystems. Cell Mol Life Sci, 2015, 72(22): 4287–4308.
[47] Singh J, Behal A, Singla N, et al. Metagenomics: Concept, methodology, ecological inference and recent advances. Biotechnol J, 2009, 4(4): 480–494.
[48] Xu B, Xu WJ, Li JJ, et al. Metagenomic analysis of thefecal microbiome reveals a broad diversity of bacterial and glycoside hydrolase profiles related to lignocellulose degradation. BMC Genom, 2015, 16: 174.
[49] Ilmberger N, Güllert S, Dannenberg J, et al. A comparative metagenome survey of the fecal microbiota of a breast- and a plant-fed Asian elephant reveals an unexpectedly high diversity of glycoside hydrolase family enzyme. PLoS ONE, 2014, 9(9): e106707.
[50] Gilbert JA, Dupont CL. Microbial metagenomics: beyond the genome. Ann Rev Mar Sci, 2011, 3(3): 347–371.
[51] Demaneche S, David MM, Navarro E, et al. Evaluation of functional gene enrichment in a soil metagenomic clone library. J Microbiol Methods, 2009, 76(1): 105–107.
[52] Dai X, Zhu YX, Luo YF, et al. Metagenomic insights into the fibrolytic microbiome in yak rumen. PLoS ONE, 2012, 7(7): e40430.
[53] Thornbury M, Sicheri J, Slaine P, et al. Characterization of novel lignocellulose-degrading enzymes from the porcupine microbiome using synthetic metagenomics. PLoS ONE, 2019, 14(1): e0209221.
[54] Wilhelm RC, Singh R, Eltis LD, et al. Bacterial contributions to delignification and lignocellulose degradation in forest soils with metagenomic and quantitative stable isotope probing. ISME J, 2019, 13(2): 413–429.
[55] Hess M, Sczyrba A, Egan R, et al. Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science, 2011, 331(6016): 463–467.
[56] Mukherjee S, Seshadri R, Varghese NJ, et al. 1, 003 reference genomes of bacterial and archaeal isolates expand coverage of the tree of life. Nat Biotechnol, 2017, 35(7): 676–683.
[57] Bashiardes S, Zilberman-Schapira G, Elinav E. Use of metatranscriptomics in microbiome research. Bioinform Biol Insights, 2016, 10: 19–25.
[58] Mallick H, Ma SY, Franzosa EA, et al. Experimental design and quantitative analysis of microbial community multiomics. Genome Biol, 2017, 18: 228.
[59] Carini P, Marsden PJ, Leff JW, et al. Relic DNA is abundant in soil and obscures estimates of soil microbial diversity. Nat Microbiol, 2016, 2: 16242.
[60] Sim M, Kim J. Metagenome assembly through clustering of next-generation sequencing data using protein sequences. J Microbiol Methods, 2015, 109: 180–187.
[61] Ferrer M, Beloqui A, Golyshin PN. Microbial metagenomes: moving forward industrial biotechnology. Journal of Chemical Technol Biotechnol, 2007, 82(5): 421–423.
[62] Gruninger RJ, Nguyen TTM, Reid ID, et al. Application of transcriptomics to compare the carbohydrate active enzymes that are expressed by diverse genera of anaerobic fungi to degrade plant cell wall carbohydrates. Front Microbiol, 2018, 9: 1581.
[63] Janusz G, Mazur A, Wielbo J, et al. Comparative transcriptomic analysis ofrevealed differential expression of genes engaged in degradation of various kinds of wood. Microbiol Res, 2018, 207: 256–268.
[64] Peng M, Aguilar-Pontes MV, Hainaut M, et al. Comparative analysis of basidiomycete transcriptomes reveals a core set of expressed genes encoding plant biomass degrading enzymes. Fungal Genet Biol, 2018, 112: 40–46.
[65] Emerson JB, Adams RI, Román CMB, et al. Schrödinger’s microbes: Tools for distinguishing the living from the dead in microbial ecosystems. Microbiome, 2017, 5: 86.
[66] Sorek R, Cossart P. Prokaryotic transcriptomics: a new view on regulation, physiology and pathogenicity. Nat Rev Genet, 2010, 11(1): 9–16.
[67] Giannoukos G, Ciulla DM, Huang K, et al. Efficient and robust RNA-seq process for cultured bacteria and complex community transcriptomes. Genome Biol, 2012, 13: R23.
[68] Roume H, Muller EEL, Cordes T, et al. A biomolecular isolation framework for eco-systems biology. ISME J, 2013, 7(1): 110–121.
[69] Knight R, Vrbanac A, Taylor BC, et al. Best practices for analysing microbiomes. Nat Rev Microbiol, 2018, 16(7): 410–422.
[70] Wang RR, Seyedsayamdost MR. Opinion: Hijacking exogenous signals to generate new secondary metabolites during symbiotic interactions. Nat Rev Chem, 2017, 1(3): 0021.
[71] Mayall P, Pilbrow V. Generalized Procrustes analysis of an ontogenetic series of modified crania: Evaluating the technique of modification in the Migration Period of Europe (4th–7th century AD). Am J Phys Anthropol, 2018, 166(1): 156–169.
[72] Boulesteix AL, Strimmer K. Partial least squares: a versatile tool for the analysis of high-dimensional genomic data. Brief Bioinformat, 2007, 8(1): 32–44.
[73] Witten DM, Tibshirani RJ. Extensions of sparse canonical correlation analysis with applications to genomic data. Stat Appl Genet Mol Biol, 2009, 8(1): 28.
[74] Wilms I, Croux C. Robust sparse canonical correlation analysis. BMC Syst Biol, 2016, 10(1): 72.
[75] Mchardy IH, Goudarzi M, Tong MM, et al. Integrative analysis of the microbiome and metabolome of the human intestinal mucosal surface reveals exquisite inter-relationships. Microbiome, 2013, 1: 17.
[76] Weiss S, van Treuren W, Lozupone C, et al. Correlation detection strategies in microbial data sets vary widely in sensitivity and precision. ISME J, 2016, 10(7): 1669–1681.
[77] Alessi AM, Bird SM, Oates NC, et al. Defining functional diversity for lignocellulose degradation in a microbial community using multi-omics studies. Biotechnol Biofuels, 2018, 11: 166.
[78] Hassa J, Maus I, Off S, et al. Metagenome, metatranscriptome, and metaproteome approaches unraveled compositions and functional relationships of microbial communities residing in biogas plants. Appl Microbiol Biotechnol, 2018, 102(12): 5045–5063.
Microbial degradation of lignocellulose
Congfeng Xu, Shiqi Ai, Guinan Shen, Yuan Yuan, Lei Yan, and Weidong Wang
,,,163319,,
Lignocellulose is widely found in the nature. The highly efficient degradation of lignocellulose requires synergistic interactions of varieties of microorganisms. The mechanism of synergistic interaction relationship is not entirely clear because it needs multitudinous microorganisms to participate in the process of lignocellulose degradation. With the development of microbial molecular biology and omics technology, some new methods will be provided for the research on the mechanism of microbial synergistic degradation of lignocellulose. Our previous research found that the bacterial composite microbial system shows strong degradation ability of lignocellulose at 50 °C. The consortium is composed of cultured and uncultured bacteria, but the former has no degradation ability. Metagenomics and metatranscriptomics show that the expression levels of some genes related to lignocellulosic degradation change significantly. It is possible to explain the microbiological and enzymatic mechanisms of lignocellulosic degradation by microorganisms through omics in the future. The research progress of lignocellulose microbial degradation is reviewed from the aspects of enzyme, pure culture strain, and microbial consortium. The current situation and application prospect of omics technology in analyzing the function mechanism of microbial consortium are also introduced, to provide reference for exploring synergistic interactions of lignocellulose microbial degradation.
lignocellulose, microbial consortium, synergy, mechanism of degradation
June12, 2019;
July31, 2019
National Key Research Program and Development Plan (No. 2018YFD0800906-03), Key Project of Heilongjiang Natural Science Foundation (No. ZD2018005), The Local Science and Technology Development Program Supported by the Central Government (No. ZY16A06-02), Program of Science and Technology Innovation Team in Heilongjiang Province (No. 2012TD006), Research and Development Plan of Heilongjiang Agricultural Company (No. HNK135-04-08), Support Program of Scientific Research Team and Platform of HBAU (No. TDJH201809).
Weidong Wang. Tel/Fax: +86-459-6819298; E-mail: wwdcyy@126.com
国家重点研发计划 (No. 2018YFD0800906-03), 黑龙江省自然科学基金重点项目(No. ZD2018005),中央财政支持地方发展科技项目(No. ZY16A06-02),黑龙江省高校科技创新团队项目(No. 2012TD006), 黑龙江农垦总局科技攻关项目(No. HNK135-04-08),黑龙江八一农垦大学科技创新团队项目(No. TDJH201809) 资助。
2019-08-06
http://kns.cnki.net/kcms/detail/11.1998.Q.20190805.1058.002.html
许从峰, 艾士奇, 申贵男, 等. 木质纤维素的微生物降解. 生物工程学报, 2019, 35(11): 2081–2091.
Xu CF, Ai SQ, Shen GN, et al. Microbial degradation of lignocellulose. Chin J Biotech, 2019, 35(11): 2081–2091.
(本文责编 郝丽芳)
