基于R语言的前列腺癌样本的关键基因数据挖掘
2019-12-05孙泽坤袁钱图胡建新
孙泽坤 袁钱图 胡建新
摘 要:为寻找前列腺癌组织与正常前列腺组织的关键基因,从Gene Expression Omnibus(GEO)数据库下载前列腺癌样本基因表达谱数据集GSE69223。进行芯片标准化处理后设置阈值|log2(FC)|>2且pvalue<0.05筛选出差异表达的基因,选择其中高表达的41个基因进行GO和KEGG分析,得出8个关键基因:FFAR2、THBS4、TRPM4、CLDN3、CLDN8、HPN、PLA2G2A和FOLH1基因。再经UALCAN生存分析的到3个上调后患者生存可能性降低的基因:FFAR2、HPN和FOLH1。得出的8个关键基因主要富集在细胞趋化性、细胞-细胞连接、脂肪酸代谢等通路,这些通路与前列腺癌的发生发展有着密切联系。除文献已经报道的与前列腺癌有密切联系的基因外,研究推测:CLDN3、CLDN8和FFAR2基因可能与前列腺癌特别是处于T2、T3分期的前列腺癌有着潜在的联系。
关键词: R语言;数据挖掘;前列腺癌;关键基因
【Abstract】 To search for key genes in Prostate Cancer tissues and normal prostate tissues, the gene expression profile data set GSE69223 of Prostate Cancer samples is downloaded from the Gene Expression Omnibus (GEO) database. After the chip standardization treatment, the threshold ∣log2(FC)∣>2 and pvalue<0.05 are used to screen out the differentially expressed genes, and 41 genes with high expression are selected for GO and KEGG analysis, and 8 key genes are obtained: FFAR2, THBS4, TRPM4, CLDN3, CLDN8, HPN, PLA2G2A and FOLH1 genes. After UALCAN survival analysis, the genes with reduced survival probability after three up-regulations are: FFAR2, HPN and FOLH1. The eight key genes are mainly enriched in cell chemotaxis, cell-cell junction, and fatty acid metabolism. These pathways are closely related to the development of Prostate Cancer. In addition to the genes already reported in the literature that are closely related to Prostate Cancer, it is hypothesized that the CLDN3, CLDN8, and FFAR2 genes may be potentially associated with Prostate Cancer, particularly Prostate Cancer at T2 and T3 stages.
【Key words】 R language; data mining; Prostate Cancer; key genes
0 引 言
前列腺癌(Prostate Cancer,PCa)是男性常见恶性肿瘤之一,多发于老年男性,同时具有高转移性,且早期没有明显症状,发现可能已经是晚期[1]。据美国癌症协会估计,2018年美国有大约164 690例新发PCa病例。同年大约有29 430例死于该病,这使其在世界致癌诱因统计榜单中已排至第二位[2]。与大多数其它癌症一样,PCa病情的发展取决于其扩散,因此局部疾病患者的5年生存率几乎为100%,癌症转移患者的生存率将下降至28%[3]。中国前列腺癌发病率虽远低于欧美国家,但随着中国社会老龄化程度的逐渐提高、饮食结构及生活习惯的不断改变、诊疗水平及生产工艺的亟待改进等因素,中国前列腺癌的发病率也有逐年上升的趨势[4]。
研究可知,R语言是由Ihaka和 Gentleman教授联合开发的一种计算机语言[5],现已经主要应用于数据处理、统计计算、数学建模、数据可视化等多个领域,是一款开源、免费、自由的面向对象的编程软件,并已拥有Linux、(Mac)OS X、Windows等多个版本。R语言使用的拓展包(packages)可根据用户需要自由开发,同时还可供使用者免费下载[6]。随着计算机技术及高通量测序技术的发展,生物芯片已然成为临床样本分析的一种有效方法,为疾病预测、分子诊断、新药开发发挥着强有力的助益作用[7-8]。本研究采用了基于R语言的芯片分析方法来研究前列腺癌与正常前列腺组织之间的基因差异,从GEO数据(https://www.ncbi.nlm.nih.gov/geo/)下载基因表达谱数据集GSE69223后对样本进行质量检测,数据清洗后设定阈值∣log2(FC)∣>2, pvalue<0.05,筛选出差异表达基因(FC:fold change 基因倍数变化),对其中的上调基因进行KEGG和GO分析以及UALACN(http://ualcan.path.uab.edu/)生存分析,从而发现了一些前列腺癌、特别是处于T2、T3分期的前列腺癌的关键基因,对研究前列腺癌的分子诊断、抗前列腺癌药物候选靶点提供了有益参考。
1 材料与方法
1.1 材料
芯片数据集GSE69223及芯片平台数据GPL570从GEO数据库(https://www.ncbi.nlm.nih.gov/geo/)下载得到,R语言版本为R3.6。除内置程序包外,其余拓展包下载自https://cran.r-project.org/及http://bioconductor.org/packages。
1.2 实验方法
1.2.1 数据获取及数据清洗
GSE69223基因表达谱芯片数据由美国Affymetrix公司制作,使用芯片平台为GPL570。数据集GSE69223包括15个正常前列腺组织样本以及15个前列腺癌组织样本。下载txt格式的原始数据,使用R语言获取表达矩阵、分组信息、表型数据,過滤掉没有基因名对应的探针以及对应某个基因名的多个探针。
1.2.2 聚类分析和PCA分析
使用R语言中的dist和hclust函数对30个样品进行聚类分析,初步判断15个正常样本与15个前列腺癌样本的差异,用以检测该数据集是否具有数据挖掘的潜力。再对样本进行主成分分析(PCA),用以判断是否有潜在因子影响两者之间的差异性。
1.2.3 获得表达差异基因
用T检验获得包含基因名、LogFC、pvalue等信息的数据框,以∣log2(FC)∣>2,pvalue<0.05为阈值筛选出差异基因,并规定LogFC>2为上调,LogFC<-2为下调。
1.2.4 差异表达基因的KEGG分析和GO分析
使用R语言中的clusterProfiler包对差异表达基因中的上调基因进行KEGG分析和GO富集分析。找出该基因的功能和富集的KEGG信号通路等信息。
1.2.5 生存分析
将经KEGG分析和GO分析的上调差异基因上传到UALACN(http://ualcan.path.uab.edu/),选择prostate adenocarcinoma(前列腺腺癌)进行生存分析,获得差异基因与生存时间之间的关系。
2 结果与分析
2.1 数据获取及数据清洗
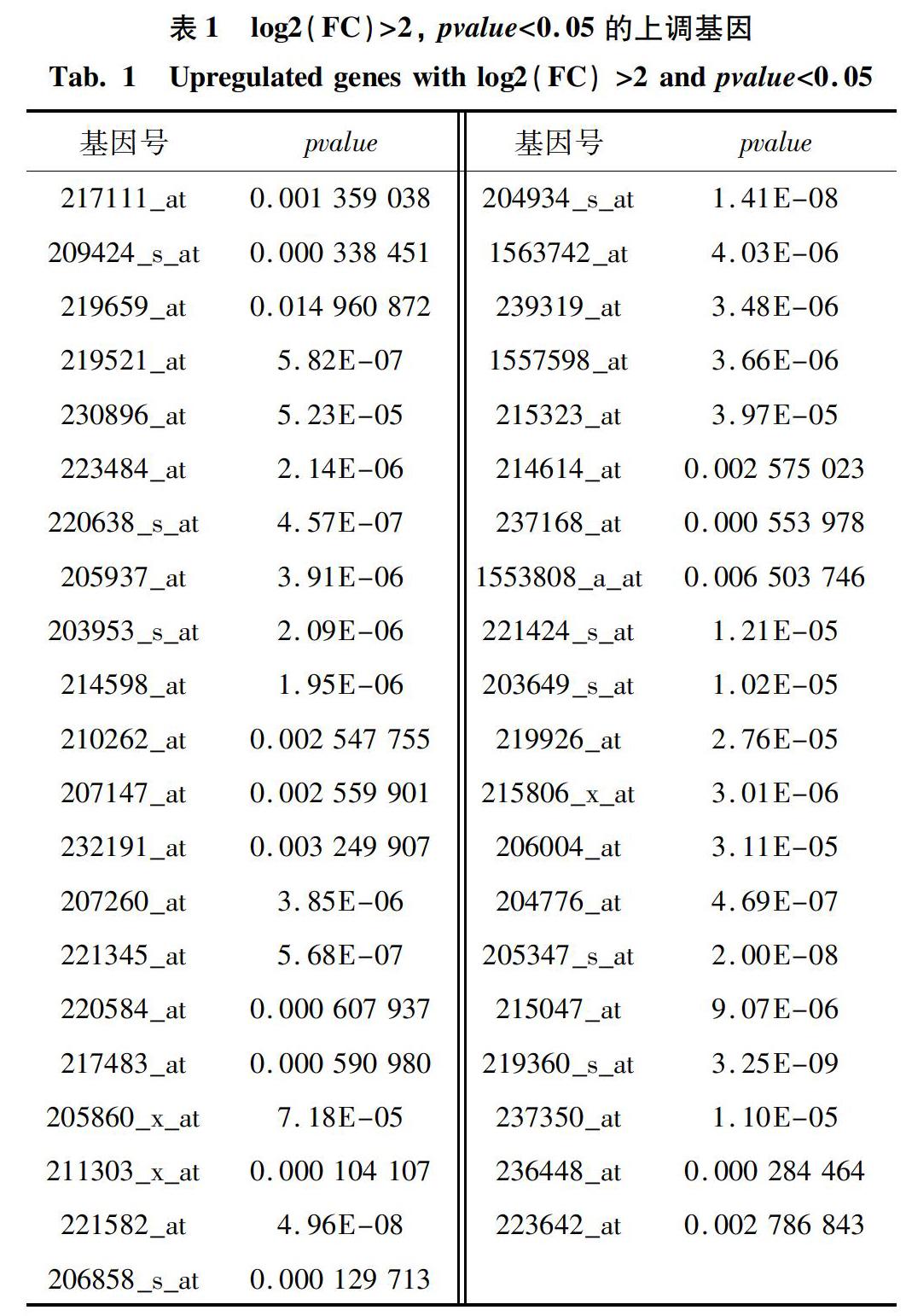
数据集包括15个正常前列腺样本以及15个前列腺癌样本的、共54 675个基因。通过数据清洗及标准化过程,可得与探针具有一一对应关系的基因有23 521个。为检验基因表达量的准确性,研究绘制了管家基因(GAPDH)以及β-actin的箱型图(见图1(a)),发现两者的表达量平均值都在0附近,这表明此数据集中的基因表达未出现异常,在误差允许范围内可进行后续分析。将30个样本纳入分析范围,图1(b)展示了各样本中基因的表达情况。
2.2 聚类分析和PCA分析
为初步判断30个样本中的前列腺正常样本(normal)与前列腺癌样本的差异,研究对样本进行了聚类分析和PCA分析。分析结果表明,样本中的某些基因的差异表达,可作为前列腺癌的诊断依据。在此次聚类分析中,有10个正常样本与前列腺癌样本分开,准确度达到66.7%,但此数据集中样本总量为30个,分组数据较少,用聚类分析只能初步揭示正常样品与前列腺癌样品具有差异性(见图1(c))。进一步地,对样品进行PCA分析。结果表明,主成分1对样本差异性的贡献率为11.44%,主成分2对样本差异性的贡献率为9.87%,通过主成分1(PC1)和主成分2(PC2)可以将前列腺正常样本与前列腺癌样本较好的进行区分(见图1(d))。
2.3 获得表达差异基因
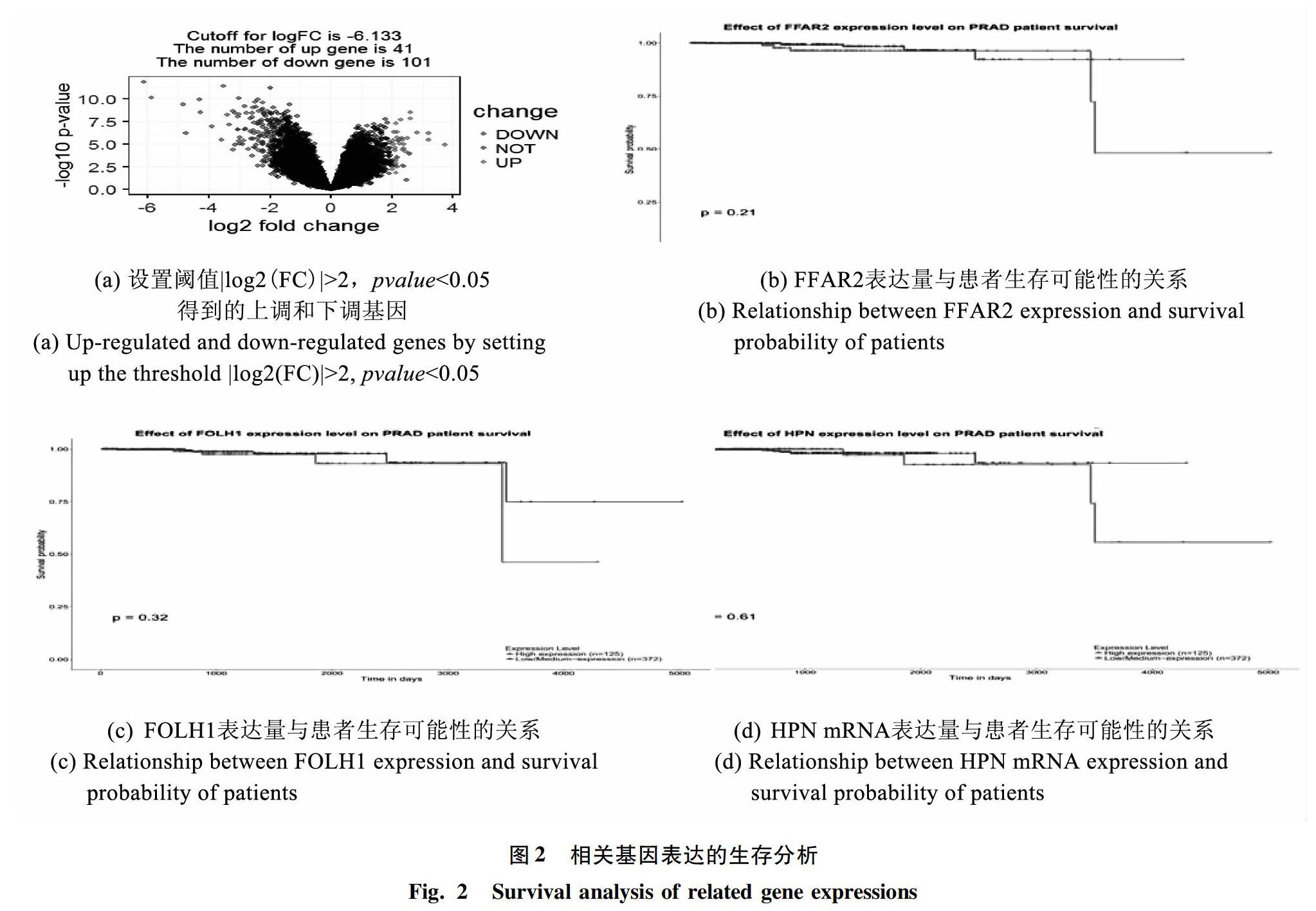
通过T检验,得到包含基因名、log2(FC)以及pvalue的数据框,设定阈值pvalue<0.05,log2(FC)>2以及log2(FC)<-2的基因,并规定log2(FC)>2的基因为上调基因,log2(FC)<-2的基因为下调基因,得到101个下调基因和41个上调基因(见图2(a))。选择前列腺癌较正常前列腺组织中的上调基因41个,导出其基因名及pvalue详见表1。
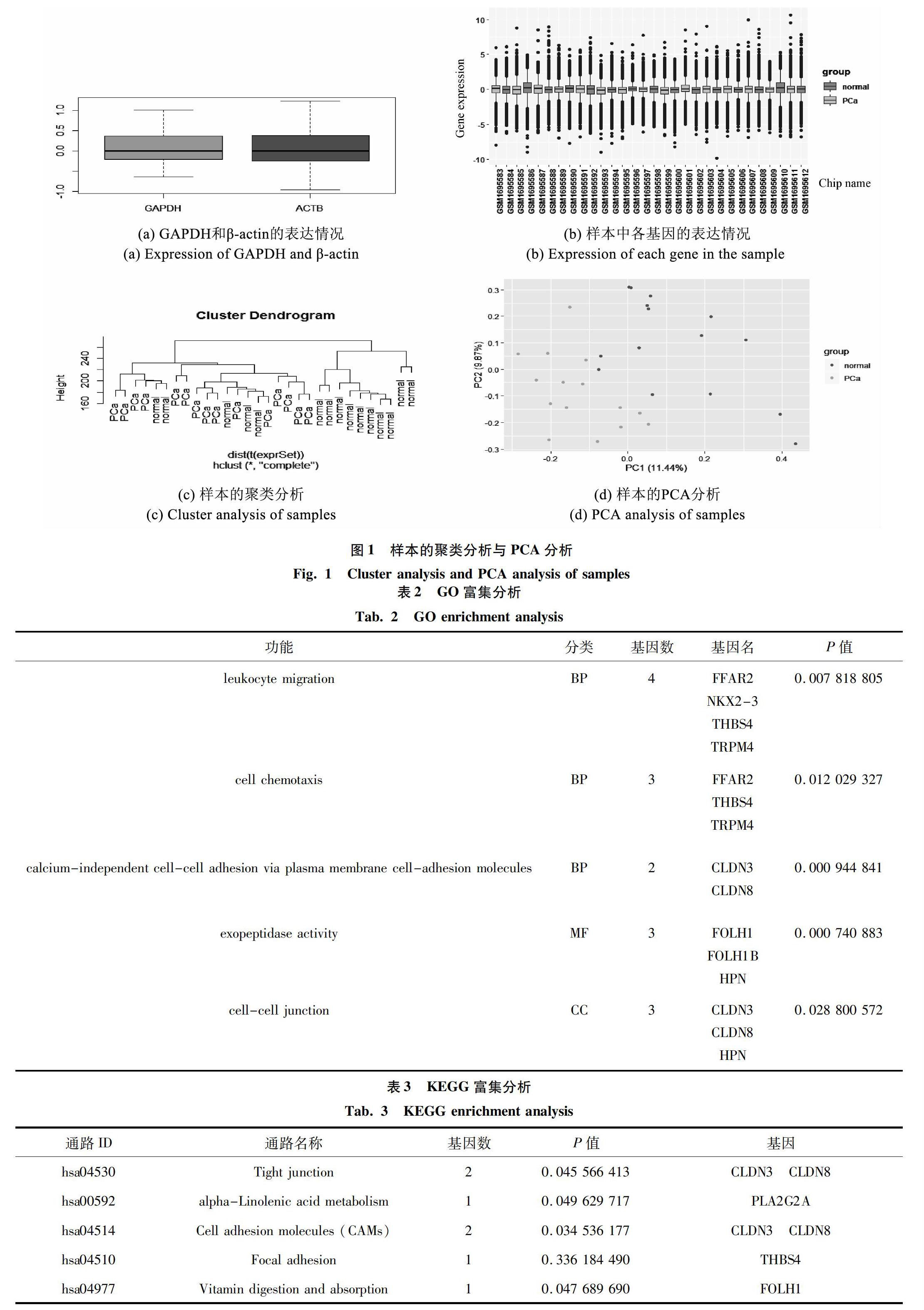
2.4 KEGG和GO分析
将得到的41个基因利用超几何分布原理在KEGG和GO数据库中进行比对,得到富集结果见表2、表3。GO分析结果表明,前列腺癌细胞较前列腺正常细胞上调的差异基因主要富集的细胞活动过程有:白细胞迁移、细胞趋化性、细胞粘附、外肽酶活性、细胞 - 细胞连接。FFAR2、THBS4、TRPM4、CLDN3、CLDN8以及HPN被富集到多条通路。FFAR2、HBS4和TRPM4三个基因与白细胞迁移与细胞趋化性有关,白细胞迁移,可能导致前列腺癌组织中的白细胞增多,白细胞产生白介素,调控多种生理生化反应。该样本中前列腺癌样本集中于T2、T3分期,该时期的前列腺癌存在转移潜能,因此可能与细胞趋化性有关。CLDN3和CLDN8是Claudin家族基因,该基因编码的蛋白由Shoichiro Tsukita及其同事在1998年发现,是细胞紧密连接的重要分子,已有报道称Claudin-1在结肠癌、Claudin-10在肝细胞癌、Claudin-18在胃癌中具有一定的临床价值[9-11]。HPN基因又叫Hepsin基因,该基因编码一种II型跨膜丝氨酸蛋白酶,该蛋白酶可能参与多种细胞功能,包括凝血和维持细胞形态。编码蛋白的表达与癌症,尤其是前列腺癌的生长和发展有关[12]。KEGG富集分析结果表明,差异表达的基因主要集中在紧密连接信号通路、多种生物分子代谢信号通路(在此列举一条α-亚麻酸代谢信号通路)、细胞粘附分子(CAMs)信号通路、黏着力信号通路、维生素消化吸收信号通路。除GO分析结果涉及的基因外,KEGG分析中还出现了PLA2G2A、FOLH1两个基因。PLA2G2A基因编码的蛋白是磷脂酶A2家族(PLA 2)的成员。该基因产物属于Ⅱ类,含有分泌型PLA 2,这是一种低分子质量的胞外酶,需要钙离子进行催化。也可催化磷酸甘油中sn-2脂肪酸酰基酯键的水解,释放游离脂肪酸和溶血磷脂,并参与生物膜磷脂代谢的调控[13]。同时,通过富集的结果来看,该基因还参与其他生物大分子如亚油酸代谢、脂肪消化吸收、醚脂代谢、花生四烯酸代谢、甘油磷脂代谢。而脂肪酸的氧化代谢过程已被证实与前列腺癌的发生和发展有着密切联系[14]。FOLH1基因编码属于M28肽酶家族的Ⅱ型跨膜糖蛋白。该蛋白以谷氨酸羧肽酶的形式存在于不同的替代底物上,包括营养叶酸和神经肽N-乙酰-1-天冬氨酰-1-谷氨酸,在前列腺、中枢神经、外周神经系统和肾脏等多种组织中均有表达。在前列腺中,该基因编码的蛋白质(PSMA)在癌细胞中被上调,并被用作前列腺癌的有效诊断和预后指标[15]。
2.5 生存分析
在UALCAN得到的生存分析结果中,研究发现,在候选的8个基因中,有5个基因的高表达组的生存可能较高,而FFAR2、FOLH1、HPN高表达组的生存可能性较低(见图2(b)~(d))。其中,已经有文献报道HPV编码的蛋白与前列腺癌有关[12],FOLH1基因编码的蛋白已成为前列腺癌的肿瘤标志物[11],而在相同的数据库相同样本的情况下,FFAR2组的P值最小,差异最为显著,因此研究推断,FFAR2基因与前列腺癌有较大关联性。
3 结束语
R语言作为一种操作简单、免费、开源的编程语言,适用于多种操作系统,为使用者提供了极大的方便。此次研究从GSE69223基因表达谱数据集中获取了54 675个基因,设定阈值pvalue<0.05,log2(FC)>2,筛选出其中的41个上调基因,并对这些基因进行了KEGG分析和GO分析,获得8个关键基因FFAR2、THBS4、TRPM4、CLDN3、CLDN8、HPN、PLA2G2A以及FOLH1。其中,FFAR2、THBS4、TRPM4三个基因与细胞趋化性相关,查看该数据集的表型数据发现,肿瘤样本全部处于T2、T3时期,此3个基因的上调,印证了该分期的前列腺癌继续发展可能转移的事实。CLDN3和CLDN8属于Claudin家族基因,该基因编码的蛋白是细胞紧密连接的重要分子,已有报道称Claudin-1在结肠癌、Claudin-10在肝细胞癌、Claudin-18在胃癌中具有一定的临床价值,因此研究推测CLDN3和CLDN8两个基因可能与前列腺癌有潜在联系。PLA2G2A参与多种脂类大分子代谢,而脂肪酸的氧化代谢过程已被证实与前列腺癌的发生发展有着密切联系。HPN和FOLH1已被文献报道与前列腺癌有着密切联系,并且FOLH1编码的蛋白(PSMA)还被用作前列腺癌的肿瘤标志物,在前列腺癌的诊断和预后中起着不可替代的作用。通过生存分析,研究还发现这8个关键基因中,FFAR2、HPN以及FOLH1三个基因的高表达会减低患者生存可能性,除文献已经报道的HPN核FOLH1基因外,本文再次经过分析推测后指出,FFAR2基因与前列腺癌的发生及发展有着潜在的关联性。但要明确其具体机制,却还需展开进一步研究。
参考文献
[1]SHI Wei, DONG Li, BAO Junsheng. Progress in the studies of prostate cancer related molecules[J]. National Journal of Andrology, 2015, 21(4):357-362.
[2]America Cancer Society. Cancer Information, Answers, and Hope[EB/OL]. https://www.cancer.org/cancer/prostate-cancer/about/key statistics.html.
[3]MILLER K D, SIEGEL R L, LIN C C, et al. Cancer treatment and survivorship statistics,2016[J]. CA Cancer J Clin. 2016,66(4):271-289.
[4]萬克松. 手术去势间断联合抗雄激素药物治疗晚期前列腺癌临床疗效研究[D]. 广州:南方医科大学, 2012.
[5]IHAKA R, GENTLEMAN R. R: A language for data analysis and graphics[J].Journal of Computational and Graphical Statistics,1996, 5(3) :299-314.
[6]韩俊伟, 智慧, 王宏, 等. R语言在生物信息实践中的应用[J]. 生物技术世界, 2015(2):180.
[7]李喜莹, 李珊珊. 生物芯片技术及其在临床检验医学中的应用进展[J]. 分子诊断与治疗杂志, 2011,3(1):62-67.
[8]于颖彦. 生物芯片在胃癌药物病理学研究中的先导作用[J]. 上海交通大学学报(医学版), 2007,27(5):491-493.
[9]李东升, 王巍, 李晴, 等. 结肠癌组织中Her-2和Claudin-1的表达及意义[J]. 广东医学, 2012, 33(2):237-239.
[10]张正东. Claudin-1和Claudin-10在肝细胞癌中的表达及意义[D]. 合肥:安徽医科大学, 2011
[11]左忠林, 陈鹏, 陈小龙,等. Claudin-18在胃癌中的临床表达关系与治疗[J]. 中华临床医师杂志(电子版), 2018, 12(3):173-176.
[12]KIM H J , HAN J H , CHANG I H , et al. Variants in the HEPSIN gene are associated with susceptibility to prostate cancer[J]. Prostate Cancer and Prostatic Diseases, 2012, 15(4):353-358.
[13]洪双双. PLAG1和PLA2G2A在肝癌中的异常表达[D]. 郑州:郑州大学, 2011.
[14]姜伟. 复杂疾病特异的基因网路与microRNA-TF协同调控网络的构建[D]. 哈尔滨:哈尔滨医科大学,2008.
[15]MARAJ B H , MARKHAM A F. Prostate-specific membrane antigen (FOLH1): recent advances in characterising this putative prostate cancer gene[J]. Prostate Cancer and Prostatic Diseases, 1999, 2(4):180-185.
