PVC-U加工特性杂谈(三)
2019-12-04王文治
王文治
(顾地科技股份有限公司,鄂州,436000)
3 “PVC凝胶化度60%到70%,性能最好”,是真的吗?
一问到PVC的凝胶化度多少最好,好多人会脱口而出:60%到70%最好。
果真如此吗?
要回答这个问题,首先要搞清楚怎么测定凝胶化度。
3.1 凝胶化度的测定
PVC的凝胶化度是指以次生微晶为交联点的三维大网络的强度,测定凝胶化度的方法有两大类,一类是测定三维大网络的强度,另一类是测定次生微晶占微晶总量的比例。
A.测定三维大网络的强度
a.溶剂吸收法
这是最简便、最常用的方法。它的原理是把被测样品浸泡在适当的溶剂中,凝胶化度越高,耐溶剂的能力就越强。常用的溶剂有二氯甲烷和丙酮,前者的方法标准为GB/T 13526—2007《硬聚氯乙烯(PVC—U)管材二氯甲烷浸渍试验方法》;后者的方法标准为 ASTM D2152-13 《Standard Test Method for Adequacy of Fusion of Extruded Poly(Vinyl Chloride)(PVC)Pipe and Molded Fittings by Aceton Immersion》。这两种方法都只是对PVC-U管材和管件的凝胶化度或熔合 (Fusion)是否达标做出评价,不能定量测定。
Fillot等[1]提出一种定量测定的方法,将一定型状、尺寸的样品浸泡在二氯甲烷中1 h,称量浸泡前后样品的重量差,样品凝胶化度越高,溶胀度越大,增重越大。但由于样品的型状、比表面等因素对测定结果影响较大,因此少有应用。
b.毛细管流变仪法
毛细管流变仪法是根据熔体通过零长毛细管时的入口压力降与熔体弹性的相关性来评价凝胶化度。此方法于1971年由A.Gonze提出[2]。
此法的要点是将待测样品的配混料在五个以上的温度造成粒料,然后,在相同条件下测定各所得粒料通过毛细管流变仪的入口压力降,将各粒料的入口压力降对其造粒温度作图,得到一S形曲线,称为参考曲线,见图1。随后,通过内扦法测定待评价样品的凝胶化度,见公式1
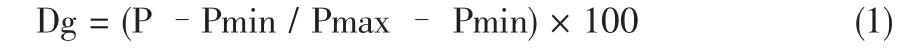
式中,Dg为凝胶化度;
P为被测样品的入口压力降:
Pmin和Pmax分别为参考曲线的最小和最大入口压力降。
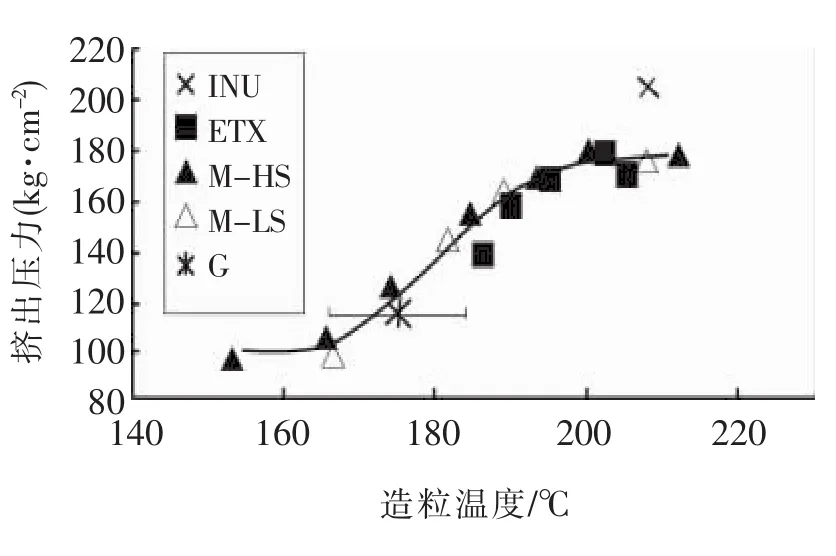
图1 参考曲线Fig.1 Reference curve
此法是最能够反映大三维网络结构强度的方法,是用于研发工作的很好方法,但需要测定多个不同温度造粒的样品的入口压力降,过程繁复,不适用于生产控制。
c.转矩流变仪法
PVC的转矩流曲线在熔合扭矩之后,由于物料温度上升(见图2曲线3)使粘度下降,扭矩随之下降(见图2曲线1)。另一方面,物料的凝胶化度随温度升高而升高[3],其走势与温度曲线相同。如果排除物料粘度随温度的升高而下降这一因素,则可以认为扭矩曲线的走势应该与温度曲线相同,这一假设可虚拟为镜面扭矩曲线,见图2中曲线2,这就揭示了使用转矩流变仪评价凝胶化度的可能性。
基于上述的推测,王文治等[4]在转矩流变曲线的最小扭矩a,熔合扭矩c,熔体扭矩e,以及两个中间点b和d处(见图2)停机取样,将样品快速投入冰水中冷却,然后破碎并测出这5个样品的转矩流变曲线,结果见图3。
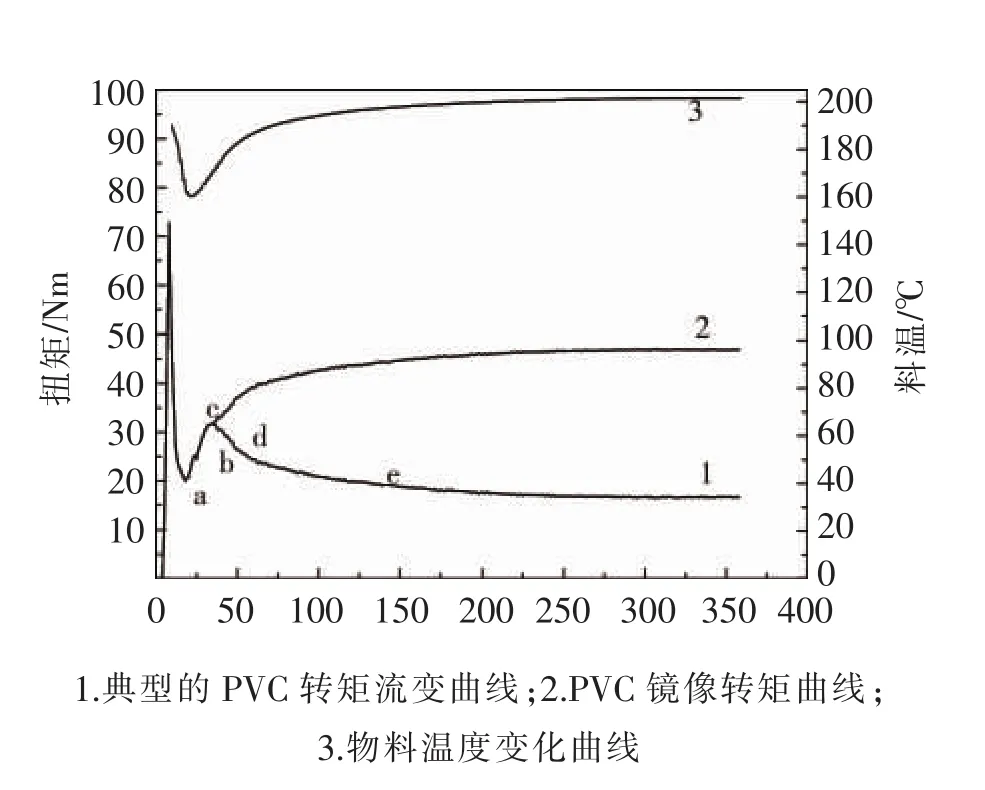
图2 PVC转矩流变曲线上的取样点Fig.2 Sampling point on PVC torque rheological curve
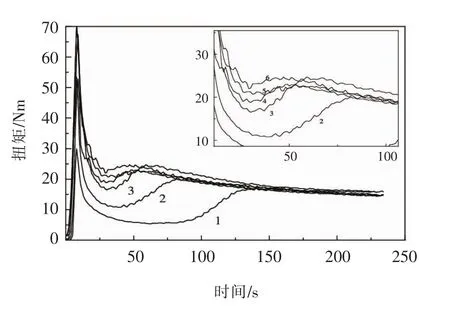
图3.a,b,c,d,e点处样品的转矩流变曲线(2~6)以及参比曲线(1)Fig.3 Torque rheological curve(2-6)and reference curve(1)of the sample at points a,b,c,d and e
从图3看出,随着样品凝胶化度的增高,流变曲线的最小扭矩越来越接近最大扭矩,说明样品的凝胶化度越高,最大扭矩与最小扭矩的差值越小。
将手工混合的配混料作为参比配混料,因未经受剪切和加热,其凝胶化度为零。将最大扭矩与最小扭矩两者的差值为零的样品,定义其凝胶化度为100%,则用式(2)可计算出被测样品的凝胶化度Dg。
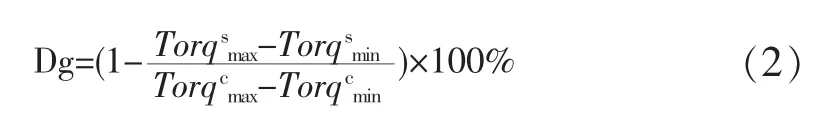
最小扭矩值随凝胶化度的变化,实质上是定量地表征了凝胶化过程PVC颗粒层次结构的变化和由此引起的粘弹性的变化,这和毛细管流变仪法测定凝胶化度的原理是相同的[5]。这个方法只需作出样品和参比配混料的转矩流变曲线,操作比较简便。与毛细管流变仪法相同,这一方法只能用于评价同一种配混料,在不同加工条件的作用下,制品凝胶化度不同。
这一方法虽然已制订为国家标准 [GB/T 34917—2017硬聚氯乙烯(PVC—U)制品凝胶化度的测定 转矩流变仪法],但其实际应用还有待进一步完善。
B.测定微晶的变化
a.差示扫描量热法(DSC)测凝胶化度
通过测量次生微晶占微晶总量百分比而测量凝胶化度的方法有差示扫描量热(DSC)法,这一方法于1981年由Gilbert等提出[6]。这一方法的原理见图4。
PVC颗粒的DSC升温曲线只有一个吸热峰,这是原生微晶(PVC聚合过程生成的微晶)的熔融峰,见图4a。经历了加工过程的PVC,其DSC曲线出现二个熔融峰,见图4b,低温峰(A)为次生微晶熔融峰,高温峰(B)为加工过程中未熔融的原生微晶熔融峰,按这两个熔融峰的热焓HA和HB的比值,即可计算凝胶化度Dg,见式(3)。

b.DSC法测定加工温度Tp
用DSC法还可以同时测定PVC在加工过程中所经历的真实(最高)加工温度Tp,因为在PVC加工过程中,熔点相当或高于最高加工温度Tp的原生微晶发生退火,导致熔点轻微提高。而在Tp温度以下熔化的微晶会在冷却过程中发生重结晶,生成次生微晶,其熔点低于Tp。这就使得A峰与B峰之间出现不存在微晶的狭小温度间隙,也就是说,经历了加工过程的PVC,没有熔点处于这一狭小温度间隙之内的微晶,因此,可以用B峰的始点温度来表征PVC的真实加工温度Tp,见图4c之交叉点1。

图4 PVC升温过程的DSC曲线Fig.4 DSC curve of PVC heating process
这种方法的国际标准为 [ISO 18373-1:2007 Rigid PVC pipes—Differential scanning calorime-try(DSC)method — Part 1:Measurement of the processing temperature],并已转化为国家标准 [GB/T33466.1--2006硬聚氯乙烯管材 差示扫描量热法 (DSC)第1部分:加工温度的测量],并作为评价凝胶化度的一种方法,Tp越高,次生微晶占微晶总量的比例越高,则Dg越高。
DSC法精密度高,所需的样品量只有几十毫克,可以进行精准测定,这是它的突出优点。
3.2 几种测定方法的相关性
我们对凝胶化度感兴趣,是因为要弄明白凝胶化度这一结构参数对制品性能的影响。不同方法从不同结构层面测定PVC的凝胶化度,ISO 1452-2:2009的表9(见表1)并列出三种测定凝胶化度的方法:二氯甲烷浸泡法 (ISO 9852),DSC法测定加工温度(ISO 18373-1)以及测定管材、管件的纵向拉伸性能(ISO 6259-1和ISO 6259-2)。表中标明,二氯甲烷浸泡法是测定凝胶化度,后面两种方法是供替代的方法,如有争议,则使用二氯甲烷浸泡法。
注:ISO 1452--2 [Plastics piping systems for water supply and for buried and above-ground drainage and sewerage under pressure----Unplasticized poly(vinyl chloride) (PVC-U)—Part 2:Pipes(给水和埋地及地面上加压排水、排污的塑料管道系统---PVC-U第2部分:管材)]

表1 物理性能Tab.1 Physical characteristics
仔细阅读这个表,觉得有几个问题颇费思量,A.DSC是高精密度的方法,为什么标准中采用的是测定加工温度Tp的方法,而不是以次生微晶占微晶总量百分比的测定凝胶化度的方法。
B.拉伸性能是产品的使用性能,为什么用它来作为测定凝胶化度的替代方法。
C.二氯甲烷浸泡法不能定量测试凝胶化度,而两个替代方法是定量测试,为什么有争议时,还是以二氯甲烷浸泡法说了算呢?
虽然无从直接找到答案,不过仔细思考,似乎可以觉察到标准制订者们的良苦用心。
ISO 1452-2:2009 于 2009 年发布,ISO 18373-1:2007则是于2007年发布,说明这两个标准是密切关联的。ISO 18373-1:2007的引言中提到“以后相关产品标准中,可能会加入规定B峰起始点或最高加工温度的要求”。这说明制订ISO 18373-1:2007,就是为ISO 1452-2:2009准备的,这是因为控制凝胶化度对于PVC承压管材的加工过程很重要,而真实加工温度Tp是影响PVC管材凝胶化度的最重要工艺参数。
另一方面,控制凝胶化度的目的是为了控制产品的使用性能,拉伸屈服强度反映管材的刚性,断裂伸长率反映管材的韧性。屈服强度太低,管材在水压作用下容易鼓胀,继而破裂;断裂伸长率太低,则管材会发生脆性断裂。所以选用了ISO 6259测定PVC管材拉伸性能,以间接表征管材的凝胶化度。
虽然DSC和拉伸性能都可以定量,但只能间接地表征凝胶化度,而三种方法中真正表征凝胶化度的,还只是二氯甲烷浸泡法,尽管它不能定量,而至今又还未能找到这三种方法之间的相关数量关系,所以,当三种方法的结果不一致时,还是以二氯甲烷浸泡法为准。
这就是测定凝胶化度目前所处的无奈境况!
3.3 凝胶化度对制品性能的影响
最早报道凝胶化度与制品性能关系的是Benjamin[7]于1980年发表的论文。
他在文章开头指出,人们根据生产PVC-U管材的多年经验,认为随凝胶化度增加,
管材强度和刚性升高并达到最大值,而韧性则达到峰值后下降。为验证这一认识,他用K值65的PVC树脂,典型的管材配方,制取4个管材样品,用毛细管流变仪法测得样品的凝胶化度分别为32%,44%,68%和90%。4个样品的力学性能测试结果见表2,从表2可见,拉伸屈服强度随凝胶化度的增加而提高,在凝胶化度为68%和90%处达到最高值,断裂伸长率在凝胶化度为44%时呈最大值。0℃的拉伸冲击强度和断裂伸长率在凝胶化度为68%处达最大值;20℃的拉伸冲击强度和断裂伸长率在凝胶化度为44%处达最大值。

表2 PVC-U管材凝胶化度对物理性能的影响Tab.2 Influence of gelation degree of PVC-U pipe on physical properties
对这4种凝胶化度的管材做20℃和60℃的1000 h水压试验,凝胶化度32%的试样,60℃的水压试验仅仅在10~15 h就发生脆性破裂。凝胶化度44%的样品,60℃的首次脆性破裂时间接近300 h,而20℃的试验没有观察到脆性破裂。凝胶化度68%和90%的样品,60℃试验在1000 h之前没有岀现脆性破裂,其20℃的试验曲线能够线性外推到25 N/mm2的特性值,因而可以认为,其出现脆性破裂的可能性会超过50年。
其后Marshall等[8]于1983年发表文章,报道他们研究了三种配混料的挤出片材的口模温度和用毛细管流变仪法测得的熔合(凝胶化)程度对冲击强度的影响,发现冲击强度随口模温度升高或随熔合程度升高都呈现峰值,见图5和图6,当熔合程度为60%~70%时,冲击强度达最大值。
关于凝胶化度对制品性能的影响,Summers等[9~11]有另外一种观点,认为加工温度升高使抗冲击性能下降,不是因为凝胶化度增高造成的,而是因为润滑失效,熔体破裂造成的。
他们使用异向锥形双螺杆挤出机挤出壁板型材,配方很简单:PVC 100份,二丁基锡稳定剂4份,硬脂酸钙1份,石蜡1份。虽然也观察到冲击强度随挤出熔体温度的升高而出现峰值,见图7,但发现随着熔体温度升高发生熔体破裂,型材表面变粗糙,见图8。将样品在相应挤出温度的温度下加压抛光,使样品表面光滑,则冲击强度随熔体温度升高而单调升高,不出现峰值,见图9。
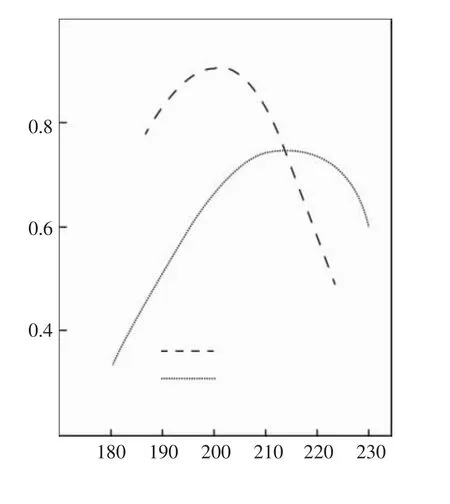
图5 口模温度对冲击强度的影响Fig.5 Effect of die temperature on impact strength

图6 冲击强度与熔合度的关系Fig.6 The relationship between impact strength and fusibility
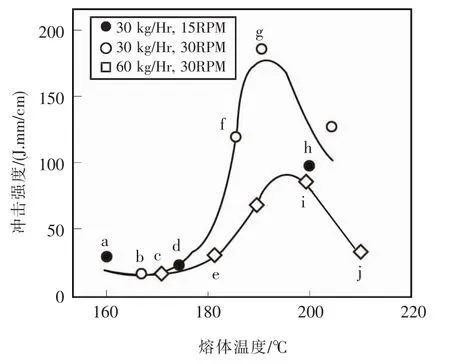
图7 韧性和挤出条件的关系Fig.7 The relationship between toughness and extrusion conditions

图8 随着熔体温度升高,样品表面呈现熔体破裂Fig.8 As the melt temperature increases,the surface of the sample shows melt fracture
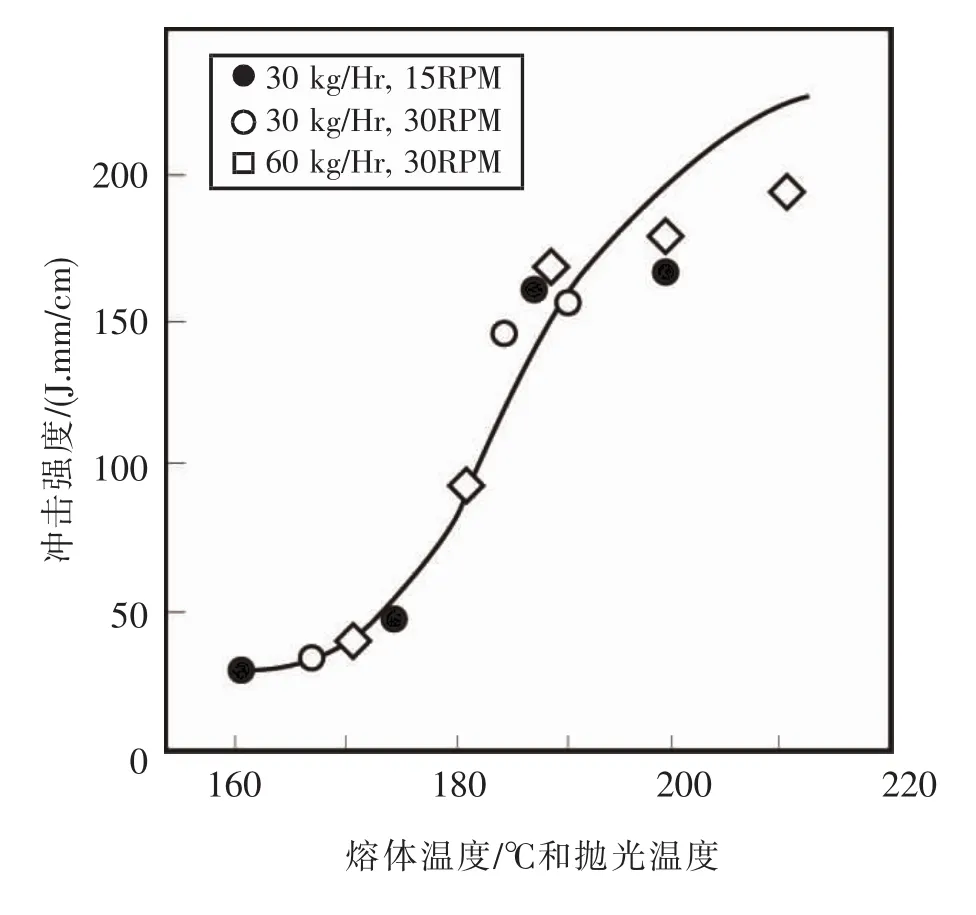
图9 加压抛光后,样品的韧性与熔体温度的关系Fig.9 The relationship between the toughness of the sample and the melt temperature after pressure polishing
他们还使用配有X-射线散射能谱(EDX)的扫描电子显微镜,检测PVC配混料在Brabender流变曲线的最低扭矩处—料温177℃和最大扭矩处—料温201℃的样品中的硬脂酸钙的分布情况。料温177℃的样品,硬脂酸钙均匀地包覆在PVC初级粒子表面,成连续相,见图10。而料温201℃的样品,PVC成为连续相,硬脂酸钙凝缩成0.1~0.2 μm的小球,成为PVC基体中的缺陷(见图11),润滑失效,使冲击强度下降。

图10 177℃样品的断面形态,硬脂酸钙均匀地分布在PVC初级粒子的表面Fig.10 Section shape of 177℃samples,calcium stearate evenly distributed on the surface of PVC primary particles
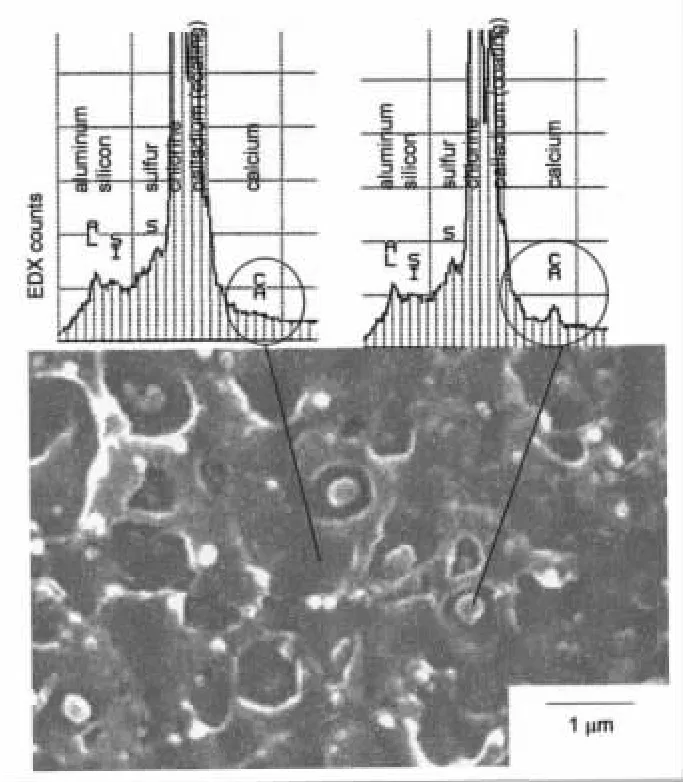
图11 201℃的样品,基体PVC上 几乎没有钙,大部份钙存在0.1至0.2um的小球上Fig.11 At 201℃,there was almost no calcium on the PVC substrate,and most of the calcium was found on the pellets of 0.1 to 0.2um
Summers等认为,加工温度升高,使三维网络强度提高,抗蠕变性能提高,抗冲击改性剂也能更好发挥效用,抗撕裂性能和长期耐压性能随之提高。但加工温度升高,致使润滑失效,熔体破裂,制品表面粗糙,抗冲击性能下降。
请回头再看看表1,也即ISO 1452-2之表9,表中列出的指标值是拉伸屈服应力≥45MPa,断裂伸长率≥80%,加工温度≥185℃,三个指标值都是只能大,不能小,这就说明ISO 2452-2所要求的是凝胶化度只能高,不能低。
3.4 DSC测凝胶化度再讨论
DSC测试方法精密度高,所需的样品只要约二十亳克,因此,可以用于对局部样品进行精准测试,这是DSC法的特出优点。
2004年VANSPEYBROECK,P等报道[12],他们为了分析管径90 mm的PVC-U管材,60℃,1000 h水压试验只有350 h就脆性破裂的原因,从管材裂口周边的外壁、芯部、内壁分别取样,见图12和图13;而对于在1000 h不破坏的管材则进行随机取样。用DSC检测所取样品的凝胶化度,测得的结果见表3和表4。

图12出现脆性断裂的管材(上边中间的黑洞为裂口)Fig.12 A pipe with a brittle fracture(the black hole in the middle of the pipe is a fissure)
图13 取样位置Fig.13 Sampling location
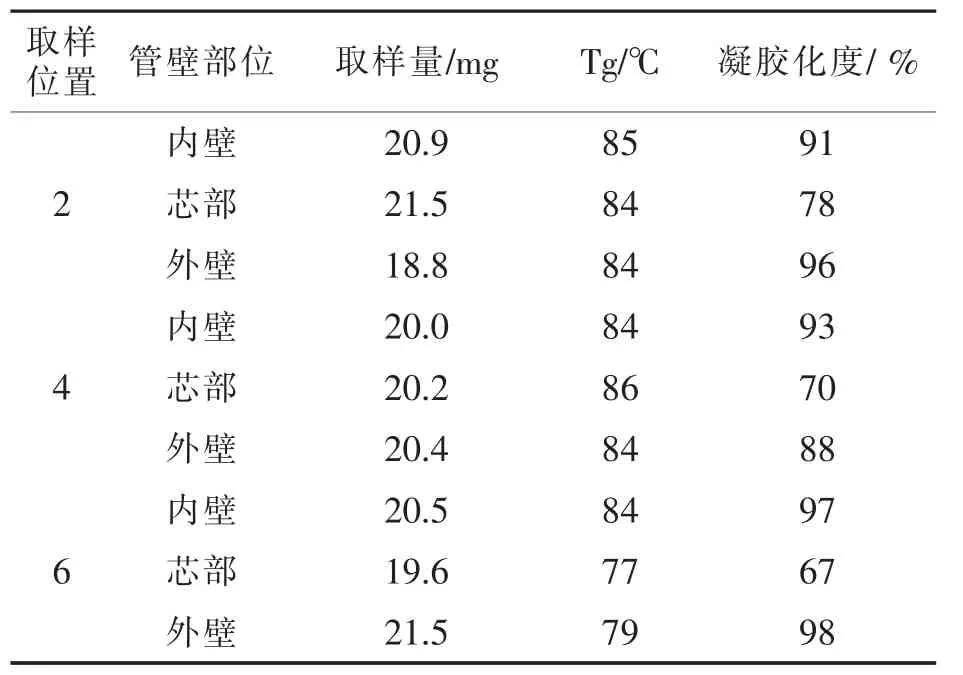
表.3出现脆性断裂的管材的凝胶化度Tab.3 The degree of gelation of a brittle fracture

表4 没有破坏的管材的凝胶化度Tab.4 The degree of gelation of tubes without damage
分析实验结果可知,不发生破裂的管材的凝胶化度都在80%以上,而且芯部凝胶化度最高,外壁最低。而350 h就发生脆性断裂的管材却是芯部凝胶化度最低,低于80%,呈现“夹生”的现象。
那么,为什么如此精准的,用百分比作为结果测定凝胶化度的方法,制订成标准方法时,变成两个非百分比为结果的方法—测定加工温度(ISO 18373-1)和测定A峰热焓 (ISO 18273-2)。而且ISO 18373-1被用在PVC-U管材产品标准ISO 1452-2:2009中时,注明这一方法不意图用于工厂的生产控制,如有争议,则应该用二氯甲烷浸泡法。
对此,可从以下相关文献中获得启示。
A.从Edwards[13]等在2014年17届国际塑料管道会议上的报告中,可找到一些答案。
Edwards等把两种PVC管材样品分别在两家实验室进行测试,并用加工温度Tp和A峰热焓对总热焓(A峰热焓+B峰热焓)的百分比两种方式处理测试结果,见表5和表6。
由表5可见,对加工温度的测定精密度很高,两家实验室之间的偏差大部分低于2%,最高为6.5%。而以百分比表示的凝胶化度,两家实验室测定结果的离散性很大,最大偏差达到81.8%,见表6。产生如此惊人的偏差的主要原因是两家实验室对DSC曲线上B峰终点温度的确定不同,实验室1确定为220℃,而实验室2确定为230℃。

表5 DSC测定加工温度Tab.5 The degree of gelation of tubes without damage
从这一实例可以看到,DSC是测定加工温度的很好方法,而要用它来以百分比表征凝胶化度,则需要有很严格的要求。这些要求包括必须使用高精密度的研究型仪器;必须配合好的软件,这样才能更好地确定B峰的终点温度,并要使B峰终点温度的确定标准化;测定时的升温速率为20℃/min或25℃/min,以避免PVC分解。还有,对样品的制备方法要有严格的规定。
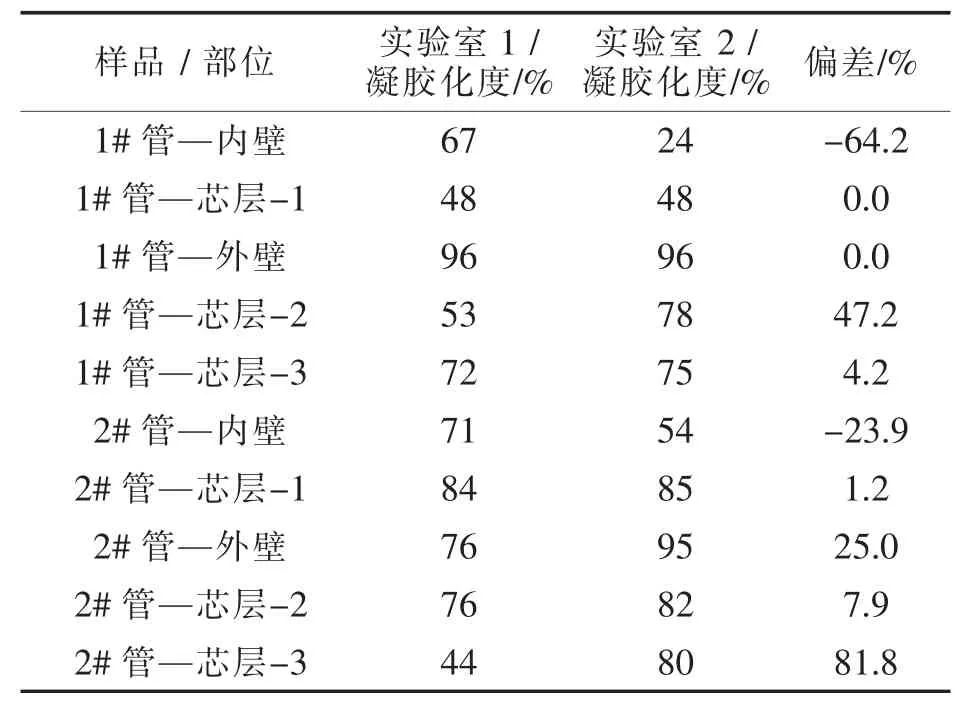
表6 DSC测定凝胶化度Tab.6 The degree of gelation was determined by DSC
B.对样品制备的要求
DSC测定的试样只需20mg,为了使这样微量试样的测定结果能够反映整体样品的特性,必须对试样的制备有严格的要求。为此,GB/T 3346.1—2016(ISO 18373—1:2007)在(试样及试验的准备)这一节规定:“分别在管材圆周的 0°、90°、180°、270°等4个方位的每个方位取至少4个样品,样品皆取自管壁的芯层。”
此外,在(结果表示)一节指出,“如果在同一根管材同一位置(如同样的角度位置)取样测得的三个独立结果之间的差异大于3℃则应进行更多的测试和/或对仪器进行重新校准。”
也就是说,对一件管材,至少要测16个样品。
因此,尽管DSC法很精准,但却不适合用于工厂的生产控制,也难以用作实验室间以百分比表示测定凝胶化度。
3.5 DSC法与三维大网络强度
20世纪90年代,欧洲的一些公司探求使用DSC法选取生产PVC-U管材、型材等产品的最佳加工温度,VANSPEYBROECK,P等[12]报道了用K值68的悬浮聚合PVC树脂,以铅盐作为稳定剂,所挤出管材的熔体温度与60℃,14 MPa环应力的水压试验时间的关系,结果见图12。

图14 熔体温度对管材破坏时间的影响Fig.14 Effect of melt temperature on the failure time of pipe
从图14看到,当熔体温度为195℃时,管材的耐水压性能最佳,高于195℃,耐水压时间下降。这就产生了一个问题,熔体温度越高,次生微晶占微晶总量的比例越高,凝胶化度越高,那么,应该抗蠕变性能越好,水压试验的破坏时间应该越长,怎么反而下降呢?反复思考,觉得DSC法把次生微晶占微晶总量的比例定为凝胶化度有失偏颇,这种定义认为只有次生微晶对三维大网络的强度有贡献,而残存的原生微晶对三维大网络的强度没有贡献,这种认识是不确当的。
PVC微晶的尺度很小,在分子链方向,微晶的厚度平均只有三个链节,而PVC含有接近10%的微晶,也就是说,对于SG 5型树脂,每根分子链平均约有30段微晶,在加工之后,有的变成次生微晶,有的仍然是原生微晶。这就是说次生微晶和原生微晶都同时存在于三维大网络当中,而且在加工过程中未熔化的原生微晶的熔点比次生微晶高,说明这些原生微晶比次生微晶更强劲,对三维大网络强度的贡献更大,换句话说,只有当原生微晶在微晶总量中占有最适当的比例时,三维大网络的强度才是最高。加工温度太高,原生微晶的比例太低,三维大网络的强度就反而下降。这就可以解释图12的结果——存在某一最佳的熔体温度,在这一温度下,原生微晶在微晶总量中占有最佳的比例,使三维大网络的强度最高,管材的耐水压性能最好。

表7实验配方Tab.7 The experimental formula
为了验证这一观点,我们使用转矩流变仪,以表7的配方,转速40 r/min,制取了不同实验温度的样品,使用转矩流变仪法测定这些样品的凝胶化度,其结果见表8。
从表8看到,对于表7的配混料,当实验温度为185℃时,样品的凝胶化度达最高值,为65.1%,此时对应的熔体温度为193.8℃。
通过反复讨论,可以归纳如下:
A.最高加工温度Tp是影响PVC凝胶化度的最重要工艺条件,DSC法能够精准地测定PVC-U制品的Tp以及次生微晶和原生微晶的比例。

表8 实验温度对凝胶化度的影响Tab.8 Effect of experiment temperature on gelation degree
B.次生微晶和原生微晶都对加工过程形成的PVC三维大网络强度有贡献,当设备和配方选定之后,存在某一最合适的Tp,这一Tp可得到最合适的次生微晶和原生微晶的比例,这一比例所构建的三维大网络强度最高,其制品的抗蠕变性能最好,表现为管材的长期承受压力的性能最优。
