西他列汀关键中间体的合成研究
2019-12-02邢冰梅徐鹤谷少华姜玉钦
邢冰梅 徐鹤 谷少华 姜玉钦

摘 要:3-氧代-4-(2,4,5-三氟苯基)-丁酸甲酯是合成二肽基肽酶-IV抑制剂类药物磷酸西他列汀的关键中间体。以2,3,5-三氟苯胺为原料,先经过重氮化反应得到1,2,4-三氟苯,再在溴素和三氯化铝的作用下发生亲电取代,得到2,4,5-三氟溴苯;然后与丙二酸二乙酯缩合,再经氢氧化钠水解、盐酸酸化、加热脱羧得到纯品2,4,5-三氟苯乙酸,最后与2,2-二甲基-1,3-二恶烷-4,6-二酮缩合后水解得到3-氧代-4-(2,4,5-三氟苯基)-丁酸甲酯,总收率达到61.9%。产品经核磁氢谱和核磁碳谱表征。该路线能有效减少副产物,适合工业化生产。
关 键 词:3-氧代-4-(2,4,5-三氟苯基)-丁酸甲酯;2,3,5-三氟苯胺;工业化生产
中图分类号:O623.6 文献标识码: A 文章编号: 1671-0460(2019)09-1930-04
Abstract: 3-Chloro-4-(2,4,5-trifluorophenyl)-methyl butyrate is a key intermediate in the synthesis of dipeptidyl peptidase-IV inhibitor cetaxidine phosphate. In this paper, 1,2,4-trifluorobenzene was synthesized from 2,3,5-trifluoroaniline by diazotization, then 2,4,5-trifluorobromobenzene was obtained by electrophilic substitution with bromine and aluminum trichloride,and then 2,4,5-trifluorophenylacetic acid was obtained via condensation reaction with diethyl malonate, hydrolysis by sodium hydroxide, acidification by hydrochloric acid and decarboxylation by heating. Finally, methyl 3-oxo-4-(2,4,5-trifluorophenyl)-butyrate was synthesized by condensation with 2,2-dimethyl-1,3-dioxane-4,6-dione and hydrolysis with a total yield of 61.9%. The products were characterized by 1H NMR and 1H NMR. This route can effectively reduce by-products and is suitable for industrial production.
Key words: 3-chloro-4-(2,4,5- three fluorophenyl)-methyl butyrate; 2,3,5-trifluoroaniline;Industrial production
作为合成西他列汀的重要中间体[1],3-氧代-4-(2,4,5-三氟苯基)-丁酸甲酯发挥着重要的作用。西他列汀(Sitagliptin,商品名Januvia)是美国Merck公司于2006年开发上市的第一种二肽基肽酶(DPP)-IV抑制剂类抗糖尿病药[2,3],主要用于Ⅱ型糖尿病的治疗[4-6]。西他列汀由于作用机制新颖,疗效确切,副作用小,已经成为临床上最畅销的治疗糖尿病药物之一,全球年销售额超过20亿美元[7],市场前景良好。其中3-氧代-4-(2,4,5-三氟苯基)-丁酸甲酯品质的好坏以及价格的高低对于西他列汀的品质及生产成本有很大的影响。因此,对3-氧代-4-(2,4,5-三氟苯基)-丁酸甲酯合成的研究具有很重要的意义。
目前,文献中报道的3-氧代-4-(2,4,5-三氟苯基)-丁酸甲酯的合成方法,主要有以下几种:
(1)起始物是1,2,4-三氟苯,在得到2,4,5-三氟苯乙酸的过程中,依次经氯甲基化、氰化、水解三个过程。再将2,4,5-三氟苯乙酸与2,2-二甲基-1,3-二恶烷-4,6-二酮缩合后水解得到3-氧代-4-(2,4,5-三氟苯基)-丁酸甲酯,该路线中氯甲基化反应步骤腐蚀性强,污染严重,且路线中需要使用剧毒氰化物,工艺具有一定的安全隐患[8]。
(2)首先需要制得格氏试剂,以2,4,5-三氟溴苯为原料,与烯丙基溴发生偶联反应后,得到2,4,5-三氟苯乙酸是通过双键氧化,再与2,2-二甲基-1,3-二恶烷-4,6-二酮缩合后水解得到3-氧代-4-(2,4,5-三氟苯基)-丁酸甲酯,该方法所用氧化剂原料价格较贵[9]。
(3)以1,2,4-三氟苯为原料,得到2,4,5-三氟苄氯是通过氯甲基化,与二氧化碳反应得到2,4,5-三氟苯乙酸,再与2,2-二甲基-1,3-二恶烷-4,6-二酮缩合后水解得到3-氧代-4-(2,4,5-三氟苯基)-丁酸甲酯,该方法中格氏试剂的制备对反应条件要求很高,并且依然存在氯甲基化三废较多的缺点[10-12]。
综合以上文献及大量实验验证,设计了一條适合工业化大生产的3-氧代-4-(2,4,5-三氟苯基)-丁酸甲酯(如图1所示),该路线以2,3,5-三氟苯胺为原料,先经过重氮化反应得到1,2,4-三氟苯,再在液溴和三氯化铝的作用下发生亲电取代反应,得到2,4,5-三氟溴苯,然后在氢化钠和溴化亚铜的作用下与丙二酸二乙酯发生缩合,再经氢氧化钠水解、盐酸酸化、加热脱羧得到2,4,5-三氟苯乙酸,最后2,4,5-三氟苯乙酸与2,2-二甲基-1,3-二恶烷-4,6-二酮缩合后水解得到目标产物3-氧代-4-(2,4,5-三氟苯基)-丁酸甲酯。该路线具有以下优点:(1)避免了剧毒氰化物的使用;(2)避免了昂贵的金属氧化物的使用;(3)避免了格氏试剂的使用,从而减少操作危险性及设备投资。
1 实验部分
1.1 试剂与仪器
酸度计(梅特勒-托利多);AV400型核磁共振仪(德国Bruker公司);LC 1260高效液相色谱仪(美国安捷伦公司)。
2,3,5-三氟苯胺(99.5%,阿尔法试剂);溴素(99%,阿拉丁试剂);三氯化铝(98%,国药集团);丙二酸二乙酯(99%,国药集团);其余试剂均为市售分析纯。
1.2 1,2,4-三氟苯的合成
在反应瓶中加入冰乙酸(500 mL)和2,3,5-三氟苯胺(150 g, 1.02 mol),降温至5~10 ℃,向体系中缓慢滴加亚硝酸钠(84.2 g, 1.22 mol)的浓硫酸(300 mL)溶液,滴加过程中保持体系温度在10 ℃以内,滴加完毕后保温反应1 h,然后继续滴加由磷酸(600 mL)、亚磷酸钠(200 g)、水(140 g)及氧化铜(15 g)配成的混合液,控制反应温度在25~30 ℃,滴加完毕后缓慢升温至50~60 ℃,5 h后,冷却,用水淬灭后乙酸乙酯(500 mL×3)萃取,最后将有机相收集,再用水(200 mL)洗涤三次,无水硫酸钠干燥、减压浓缩,得到1,2,4-三氟苯109 g,收率为81.4%。
1.3 2,4,5-三氟溴苯的合成
在反应瓶中加入1,2,4-三氟苯(100 g, 0.76 mol)和三氯化铝(10 g, 0.076 mol),室温下缓慢滴加溴素(72 g, 0.46 mol)。室温反应5 h,TLC监控原料反应完全后,用氯仿(200×3)萃取,合并有机相,用3%的稀盐酸洗涤,然后加入饱和亚硫酸钠溶液去除有机相中的溴,并依次进行水洗、饱和食盐水洗涤,蒸除溶剂得152.6 g产品,收率为96.3%。气相色谱检测纯度为99.2%(面积归一化法;色谱柱:石英毛细管柱;进样口温度160 ℃,保持1 min,按5 ℃/min升温至250 ℃,保持1 min;载气:高纯氮,流速:1.5 mL/min,进样量:1.0 μL)。
1.4 2,4,5-三氟苯乙酸的合成
将氢化钠(46 g, 1.92 mol)溶于二氧六环(500 mL)中,加热至60 ℃,在此温度下滴加丙二酸二乙酯(230 g, 1.8 mol),然后加入溴化亚铜(6.8 g, 0.048 mol),三氟溴苯(100 g, 0.48 mol),加料完毕后,将体系加热至100 ℃反应20 h。反应结束后蒸去一半溶剂,加入NaOH饱和溶液(200 mL)进行水解反应,加热回流5 h。反应结束后,蒸除溶剂,加水溶解,用甲基叔丁基醚(MTBE)萃取除去杂质。然后将水相用稀盐酸调节pH至1~2,再依次进行MTBE萃取、水洗、饱和食盐水洗、干燥、过滤、真空干燥一系列操作,得到82 g黄褐色固体,将粗品溶于异丙醚中,加热至回流,用活性炭脱色,然后趁热过滤,最后得到产品75 g,产率为83.1%,HPLC检测纯度为99.1%
1.5 2,2-二甲基-5-[2-(2,4,5-三氟苯基)-乙酰基]-1,3-二恶烷-4,6-二酮的合成
将2,4,5-三氟乙酸(50 g, 0.26 mol)溶于四氢呋喃(250 mL)中,依次加入2,2-二甲基-1,3-二恶烷-4,6-二酮(45.5 g, 0.32 mol)、N,N-二异丙基乙胺(3.36 g, 0.026 mol)和1,8-二氮杂二环十一碳-7-烯(DBU, 20 mL),将反应体系置于冰盐浴中,待体系温度降至0 ℃以下后再慢慢滴加DMSO(20 mL),滴加完毕后转移至室温,50 ℃下反应1.5 h,TLC监控反应,待原料反应完全后,加入冰饱和食盐水淬灭反应,然后用乙酸乙酯(200 mL×3)萃取,合并有机相,将有机相用饱和食盐水洗涤,干燥后浓缩,真空干燥最后得到黄色油状液体2,2-二甲基-5-[2-(2,4,5-三氟苯基)-乙酰基]-1,3-二恶烷-4,6-二酮82 g,收率98.6%。
1.6 3-氧代-4-(2,4,5-三氟苯基)-丁酸甲酯的合成
在反应瓶中,将2,2-二甲基-5-[2-(2,4,5-三氟苯基)-乙酰基]-1,3-二惡烷-4,6-二酮(50 g, 0.16 mol)加入无水乙醇(250 mL)中,然后缓慢滴加冰乙酸(9 mL),回流下反应3 h,TLC监控反应,待原料反应完全后,加入饱和食盐水淬灭反应,然后用乙酸乙酯(200 mL×3)萃取,合并有机相,将有机相用饱和食盐水洗涤,干燥后浓缩,真空干燥得到3-氧代-4-(2,4,5-三氟苯基)-丁酸甲酯37.8 g,收率为97.2%,1H NMR (600 MHz, CDCl3): δ 7.07-7.02 (m, 1H), 6.97-6.92 (m, 1H), 3.85 (s, 2H), 3.75 (s, 3H), 3.56 (s, 2H). 13C NMR (151 MHz, CDCl3): δ 171.13, 157.15, 119.77, 119.58, 105.80, 105.52, 105.31, 33.40。
2 结果与讨论
2.1 2,4,5-三氟苯胺合成反应机理路线
从1,3,4-三氟苯开始,对2,4,5-三氟苯胺的合成路线反应机理进行了分析,有助于进一步优化反应路线,反应机理路线如图2所示。
2.2 三氯化铝的量对2,4,5-三氟溴苯收率的影响
收苯环的卤代反应常常会使用Lewis酸为反应的催化剂,但由于1,3,4-三氟苯上带有三个强吸电子基团,导致苯环上的溴代较难,因此
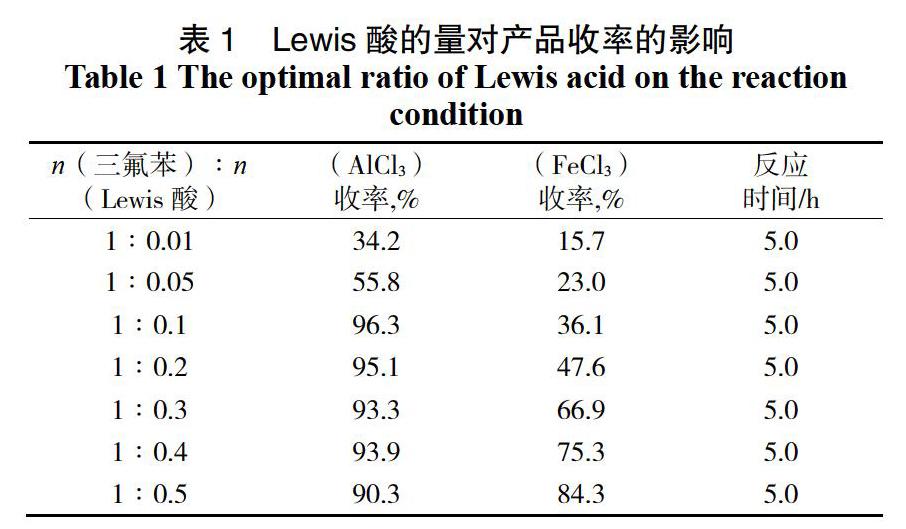
对Lewis酸的种类和量也有很严格的要求。我们固定其他反应条件不变,对Lewis酸的种类和量进行了研究(表1)。
由表1可以看出,使用AlCl3作为Lewis酸的反应效果优于使用FeCl3,并且随着催化剂量的增加,产率逐渐提高,n(1,3,4-三氟苯) : n(AlCl3)为1 : 0.1时反应收率达到最高,为96.3%,而当继续增加催化剂量时产品收率无明显提高且有逐渐下降的趋势,因此选择AlCl3作为反应的催化剂,n(1,3,4-三氟苯) : n(AlCl3) = 1∶0.1为最佳投料量比。
2.3 CuBr的量對反应收率的影响
由图2的反应机理可知,CuBr对2,4,5-三氟溴苯和丙二酸二乙酯的取代反应起到重要的催化作用,因此我们对催化剂CuBr的量进行了探讨。
由表2可知,随着催化剂CuBr量的增大,产品的收率呈正态分布逐渐提高,当n (三氟溴苯) : n (CuBr)为1 : 0.1时反应收率达到最高,为83.1%,当继续增加CuBr的量时,反应收率有下降趋势,因此选择1 : 0.1为反应的最佳摩尔比。
2.4 碱性催化剂对2,2-二甲基-5-[2-(2,4,5-三氟苯基)-乙酰基]-1,3-二恶烷-4,6-二酮合成的影响
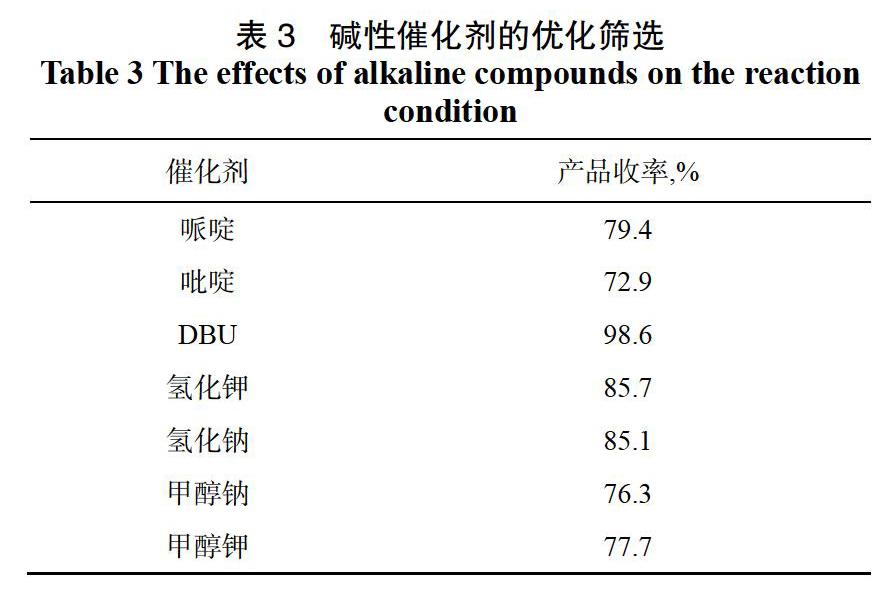
2,2-二甲基-5-[2-(2,4,5-三氟苯基)-乙酰基]-1,3-二恶烷-4,6-二酮的合成反应对碱性催化条件要求比较高,为了选择反应效果更好、反应收率更高的一种,对几种碱性化合物进行筛选(见表3)。
由表3可以看出,当使用胺类作为碱性催化剂时,反应效果不佳,产品收率较低;当使用NaH和KH作为碱性催化剂时,收率也较低,且本反应过程中有水的生成,NaH和KH遇水反应剧烈,且会产生氢气,需在低温下进行,对反应不利;当采用DBU为催化剂时,反应效果最佳,产品收率可达98.6%,且DBU对反应条件要求不苛刻。综上,选用DBU为该反应的碱性催化剂。
3 结 论
本文以2,4,5-三氟苯胺为原料合成了西他列汀的关键中间体3-氧代-4-(2,4,5-三氟苯基)-丁酸甲酯,总收率为61.9%,得到了较高的收率。分析了2,4,5-三氟苯乙酸的反应机理,探讨了2,4,5-三氟溴苯的合成中Lewis酸对其溴化反应的影响、催化剂的量及种类对缩合反应的影响,最佳反应条件得以确认。该路线原料易得、操作简便、收率高,适合工业化生产。
参考文献:
[1]胡静, 饶和平, 郑土才, 等. 2,4,5-三氟苯乙酸的合成研究进展[J]. 化学研究与应用, 2014, 26(8):1176-1182.
[2]Sbokova E, Christ A D, Boehringer M, et al. Dipeptidyl peptidase IV inhibitors: the next generation of new promising therapies for the management of type II diabetes[J]. Curr Top Med Chem, 2007, 7(6):547-555.
[3]Drucker D, Easley C, Kirkoatrick P. Sitagliptin[J]. Nat Rev Drug Discov, 2007, 6(2):109-110.
[4]夏玲红. 治疗糖尿病的新药西他列丁[J]. 中国新药杂志, 2007, 16(12):979-981.
[5]张立宏, 王莉莉, 李彬. 二肽基肽酶-IV抑制剂药理学作用及机制的研究进展[J]. 中国新药杂志, 2009, 18(11):1000-1004.
[6]Hansen K B, Hsiao Y, Xu F, et al. Highly efficient asymmetric synthesis of sitagliptin [J]. J Am Chem Soc, 2009, 131:8798-8804.
[7]Desai A A. Sitagliptin manufacture: a compelling tale of green chemistry, process intensification, and industrial asymmetric catalysis[J]. Angew Chem Int. Ed Engl, 2011, 50:1974-1976.
[8] 鲍樟水, 冯启, 夏旭建, 等. 2,4,5-三氟苯乙酸的合成[J]. 浙江化工, 2014, 45(1):4-6.
[9]Kim N D, Chang J Y, Jung J H, et al. Method for preparing intermediate of sitagliptin using chiral oxirane:WO,2011/040717[P]. 2011-04-07.
[10]Stamford A W, Gilbert E J, Cumming J N. Pyrrolidine fused thiadiazine dioxide compounds as BACE inhibitors, compositions, and their use :US,2014/023667[P]. 2014-01-23.
[11] 冯秀娟, 谭君, 杨帆, 等. 一种2,4,5-三氟苯乙酸的制备方法: CN, 101823952[P]. 2010-09-08.
[12]何人宝, 王莺妹, 金逸中, 等. 2,4,5-三氟苯乙酸的合成[J]. 化工生产与技术, 2011, 18(3):4-6.
