高通量测序技术在先天性脑积水基因诊断中的应用
2019-11-30潘蕾何志明张锐
潘蕾,何志明,张锐
脑积水是一种常见但复杂的疾病,其是由脑脊液流动的物理或功能障碍引起的,每1 000例婴儿中就有1.1例患有这种疾病[1-2]。脑积水按发病机制分为交通性脑积水和梗阻性脑积水。按病因分为由颅内出血、宫内感染等外在原因引起的继发性脑积水和无明显原因的原发性(先天性)脑积水;按照有无脑外主要临床特征可分为综合征性脑积水和非综合征性脑积水[3]。遗传性因素是先天性脑积水的重要病因,并在生育中存在较高的再发风险,是临床研究的重点。最常见的遗传原因是与L1CAM基因相关的X连锁脑积水,这只能解释5%~10%的男性病例[4]。与其他类型的脑积水相比,非综合征性先天性脑积水围生期死亡率高,发病早、病情严重、预后差,约90%的存活婴儿表现为神经和身体残疾[5]。这部分先天性脑积水无明显外在致病因素,又缺乏脑外的临床特点指导我们开展基因学检查,所以病因诊断极其困难。随基因检测技术发展,已有越来越多基因被证实或怀疑与先天性脑积水有关[6-7]。但是全外显子测序耗时长、成本高,实际难以应用于临床产前诊断,随着二代测序法(next generation sequencing,NGS)的出现,医生在临床诊疗中自主设计针对先天性脑积水的基因面板成为可能。本研究从一个8例脑积水患者家系的临床特点和遗传规律出发,尝试自主设计NGS基因面板,提高基因突变检出率,以便为这些家庭提供可靠的咨询和风险评估。
1 资料与方法
咨询者是来自深圳市宝安区妇幼保健院产前诊断门诊的1例38岁孕妇(Ⅱ2),妊娠24周超声提示胎儿(Ⅲ5)脑积水,前来就诊。该孕妇共有5次妊娠史,第一次妊娠在孕20周因彩色超声发现胎儿(Ⅲ1)脑积水引产一男婴,第二次妊娠孕24周彩色超声发现胎儿(Ⅲ2)脑积水,要求生育,孕足月“碎胎术”阴道娩出一男死婴,未行任何基因检查,随后两次妊娠均孕足月顺产一女婴,体健。脑积水家族史阳性,家系图见图1。Ⅱ11出生时头围大,出生后数天死亡。Ⅱ12出生时大拇指和食指并指内收,3个月后发现脑积水,行外科手术治疗后智力、行为接近正常,18岁时Ⅱ12失联至今。胎儿Ⅲ7于孕20周彩色超声发现重度脑积水,胎死宫内。胎儿Ⅲ12和Ⅲ13均在孕中期结构筛查超声时发现脑积水而终止妊娠。胎儿(Ⅲ5)孕24周左右彩色超声及磁共振成像(MRI)均提示重度脑积水,未发现其他伴发畸形,见图2。
本例孕妇放弃胎儿,孕25周羊膜腔注射利凡诺终止妊娠,同时行羊水染色体核型、染色体微阵列分析,引产一男死婴,头围约26 cm,双侧拇指内收,见图3(后插一)。取胎儿(Ⅲ5)皮肤组织按照标准流程提取基因组DNA(QIAamp DNA Blood Midi Kit,Qiagen,Hilden,Germany),完成单个受检者的DNA建库。自主设计包含L1CAM和AP1S2两个目标基因的基因面板,与受检者DNA库47 ℃杂交16~24 h,杂交结束后进行洗涤和洗脱反应,随后进行捕获样品的连接介导PCR反应,最后利用高通量测序仪BGISEQ-500连续双向测序50个循环,并读出原始测序数据并行序列分析。对于发现的致病突变做Sanger测序,验证基因芯片捕获和高通量测序的结果。留取本例夫妇(Ⅱ1和Ⅱ2)、家系成员Ⅱ4和Ⅱ8外周血标本行Sanger测序验证,其余成员暂未参与检测或验证。
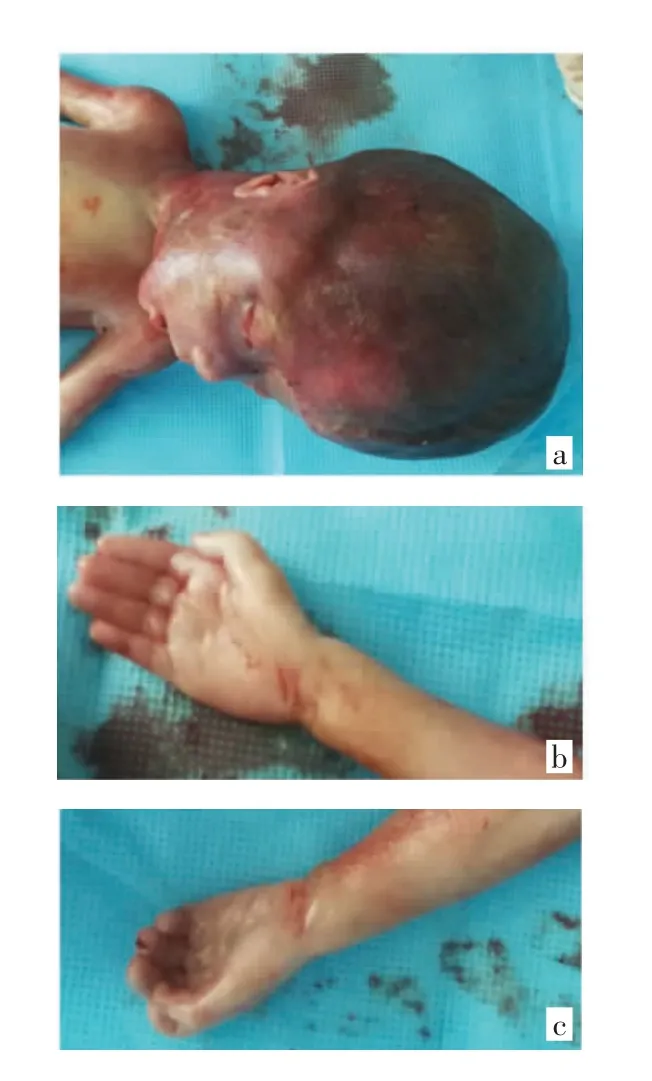
图3 脑积水胎儿Ⅲ5引产后的大体特征
2 结果
胎儿(Ⅲ5)检出L1CAM基因6号外显子突变c.551G>A(p.Arg184Gln),遗传自母亲(Ⅱ2),AP1S2基因未检测到任何突变。后续家系成员Ⅱ4和Ⅱ8取外周血标本行Sanger测序验证均为该L1CAM基因突变携带者,见图4(后插一)。
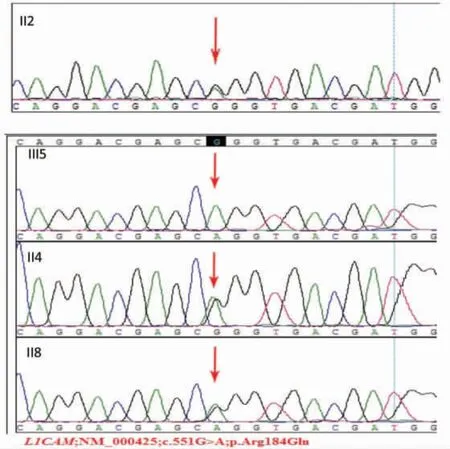
图4 家系成员L1CAM基因检测序列图
3 讨论
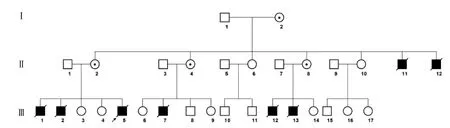
图1 X连锁脑积水家系图
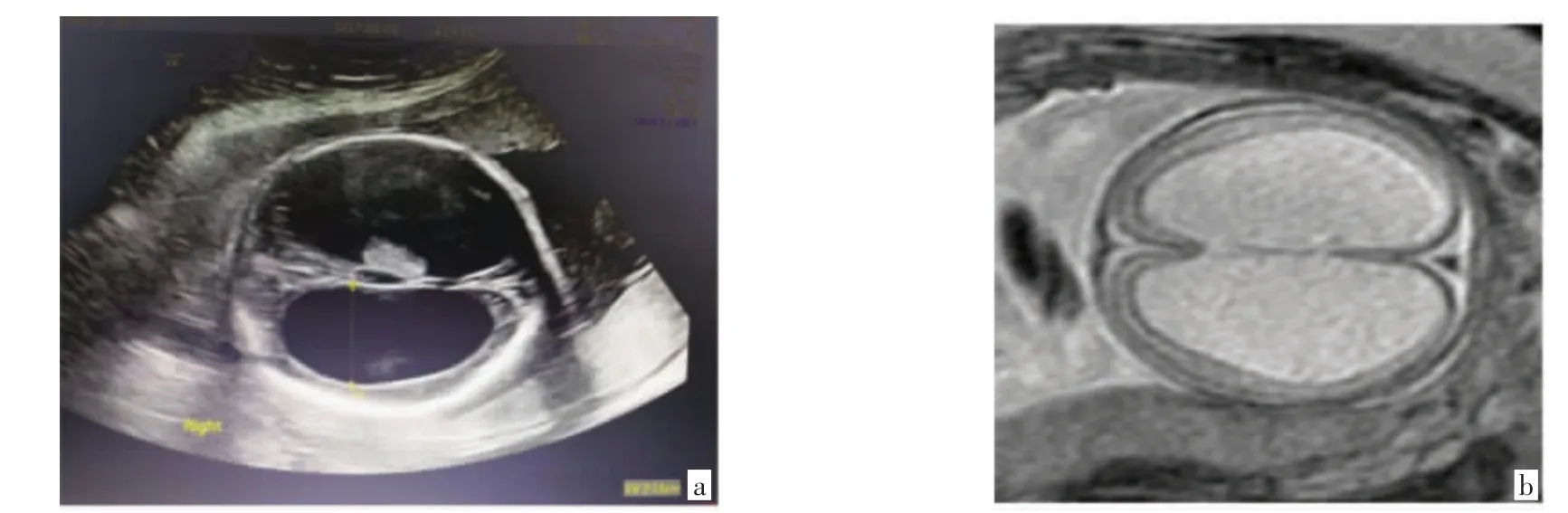
图2 脑积水胎儿Ⅲ5的影像学结果
先天性脑积水是一种严重的出生缺陷,其遗传学机制尚不清晰。相当一部分的先天性脑积水是遗传综合征的其中一种表现,而非综合征性的孤立性脑积水则较为罕见[8]。Tully等[3]综述了非综合征性先天性脑积水的4种表现形式及其相关基因,分别是X连锁脑积水伴导水管狭窄(hydrocephalus due to stenosis of the aqueduct of Sylvius,HSAS)、弗里德综合征(Fried syndrome)、肌肉-眼-脑病和严重的常染色体隐性遗传病,共涉及十余种基因。目前为止,共有4个基因与先天性脑积水关系密切,其中L1CAM和AP1S2基因突变可引起X连锁隐性遗传的先天性脑积水,而CCDC88C和MPDZ基因突变则引起常染色体隐性遗传疾病[6]。本病例家系提示X连锁隐性遗传,故设计的基因面板只含有L1CAM和AP1S2两个基因。L1CAM基因突变被认为是先天性脑积水最主要的原因。L1CAM基因突变导致了一种X连锁特征,称为L1综合征,其特点是脑积水、内收拇指、胼胝体发育不全、痉挛性截瘫和精神发育迟滞[9]。关于L1CAM基因检出率、基因型-表型关系的报道只有3个大样本研究。Finckh等[10]筛选出153例产前或临床怀疑的X连锁脑积水患者的临床标本进行L1CAM检测,共发现46个致病突变,即检出率30.1%,在102例L1综合征家族史阴性病例中有16例(16/102,15.7%)检出突变,家族史和L1病的典型临床表现数目对突变检出率有显著影响。Adle-Biassette等[11]对138例L1CAM基因筛查标本的神经病理资料进行回顾性分析,多达57例胎儿检出L1CAM突变,在性别、脑积水、内收拇指、Sylvius导水管狭窄、胼胝体异常和皮质脊髓束异常6个主要临床特征中,脑积水的阳性预测值最低44%,而内收拇指的预测值最高74%。Vos等[12]的大样本研究中内收拇指也具有最好的阳性预测价值。本病例男性、脑积水、拇指内收和多个家族成员受累这些临床特征,与之前的研究相符合。由于导水管狭窄和胼胝体情况往往因脑积水重度压迫而难以判断或得不到明确诊断,我们认为拇指内收更可能作为预测突变的标志。本次检出突变位点6号外显子c.551G>A(p.Arg184Gln)在1994—2000年被报道过6次[13-18],有3例表现为重度脑积水、1例拇指内收,余2例未提及临床表现而是从分子功能角度研究了该突变的致病机制,我们检测到的基因型基本符合其临床表型。
另一个可能导致X连锁脑积水的基因是位于X染色体的AP1S2基因,可引起弗里德综合征,首次描述于1972年[19],主要特征是智力残疾,通常伴有不同程度脑积水,如行CT扫描可见较明显的铁沉积,但产前与L1综合征区别较困难,基因诊断可能是在产前诊断中鉴别两者的主要手段[20]。
综合征性先天性脑积水由于临床特征明显,脑外畸形的形式有助于识别特定的基因状况,难度系数较小。但是对于非综合征性的先天性脑积水,除了L1综合征和弗里德综合征,可能还涉及沃克/肌肉-眼-脑疾病、纤毛病和糖基化障碍。沃克/肌肉-眼-脑疾病主要结构特征为脑积水,以及不同的眼部表现,包括微小丘脑炎、视神经发育不良和前房畸形,涉及POMT1,POMT2,POMGNT1,FKTN,FKRP,LARGE,ISPD等7个常染色体隐性遗传基因[3]。Shaheen等[6]收集先天性脑积水家系行全外显子测序,在这些家族中发现了21种可能的致病突变,涉及16个基因,其中没有一个是X连锁的。纤毛病和糖基化障碍是这项研究中最常见的先天性脑积水的病因(分别为19%和26%),涉及EML1、WDR81、CCDC88C和MPDZ等十余个基因。非综合征性先天性脑积水临床特点隐匿,故难以缩小候选基因范围,尤其对于发病例数少或者家族史阴性的病例,遗传特点分析困难,存在确诊时间长和漏诊可能。对于产前诊断这一特殊情况,不仅孕周时限性强,而且胎儿超声提供的临床特点有限,如拇指内收、智力障碍、痉挛性截瘫、肌肉或眼病很难在胎儿期做出判断,所以非综合征性先天性脑积水产前基因诊断需开发新手段、提高效率。
本例病例在产前仅有脑积水表现,未发现其他结构畸形,所以在产前阶段难以缩小候选基因的范围,而逐个可疑基因进行排查所需的时间和程序更久,漏诊可能性更大。NGS以其通量高、总体成本低、信息产出高等突出优点迅速发展,目前基于NGS的常用测序方法有Panel测序、外显组测序和全基因组测序3种[21],Panel测序可以由研究者自主设计较大的基因面板,相对价格适中、耗时短。NGS的应用为获得候选基因带来了巨大的方便,符合产前诊断的时限性要求,未来可以更高效地应用于先天性脑积水产前基因诊断。
