胞内模式识别受体在抗白色念珠菌感染中的作用研究进展*
2019-11-28余达浪陈玲霞刘志春刘志平
余达浪,陈玲霞,谢 璐,刘志春,刘志平
(赣南医学院 1.2017级硕士研究生;2.基础医学院, 江西 赣州 341000)
近年来,真菌发病率特别是白色念珠菌发病率年年上升,而且致死率也逐年增加[1-4]。免疫缺陷病人增多、抗真菌药物数量有限和耐药是造成播散性念珠菌相关疾病的高发病率和高死亡率的主要原因[5]。因此,了解宿主免疫系统如何对抗真菌感染对于开发新的基于免疫应答的疗法至关重要。模式识别受体是宿主在漫长种系进化繁衍过程中形成的对抗病原体侵袭的首要防御线[6-7]。当白色念珠菌侵入宿主后,模式识别受体(PRRs)识别并结合白色念珠菌表面的病原体相关分子模式(PAMPs),活化宿主固有免疫,从而发挥抗真菌作用[8]。宿主主要有两类PRRs[9]:一类是胞外PRRs,如Toll样受体(TLRs)和C型凝集素受体(CLRs),通过识别胞外白色念珠菌PAMPs,然后活化细胞内信号来激活宿主免疫应答;另一类是胞内PRRs,包括NOD样受体(NLRs)和RIG-I样受体(RLRs),在白色念珠菌感染的相关疾病中也发挥重要免疫调节作用。本文就胞内PRRs的类型及它们在抗白色念珠菌感染中的作用机制进行综述。
1 胞内模式识别受体的类型
目前研究发现的胞内PRRs主要是NLRs和RLRs两类。在人体内已鉴定的NLR家族分子共23种,在小鼠体内至少有34种,其中NOD1和NOD2是在NLR家族中最早报道发现的[10]。NLR家族成员中,有些可直接发挥作用,有些可以形成多蛋白复合物从而发挥作用,这种蛋白复合物被称为炎症小体(或炎性小体),包括NLRP3、NLRC4、NLRP1等。NLR家族成员中,具有抗白色念珠菌作用的主要有NLRP3、NLRC4和NLRP10。RLRs是胞内识别PAMPs的重要分子家族,包括维甲酸诱导基因I(RIG-I)、黑色素瘤分化相关蛋白5(MDA-5)、遗传学和生理学实验室蛋白2(LGP2)等分子,但有关胞内PRRs在抗白色念珠菌感染作用研究报道较少。
2 胞内PRRs在抗白色念珠菌感染中的作用
2.1NLRP3在抗白色念珠菌感染中的作用NLR通过识别白色念珠菌PAMP而活化炎症小体诱导免疫应答杀灭白色念珠菌,其中目前最广为研究的炎症小体是NLRP3[11]。NLRP3炎症小体分布在免疫细胞的胞浆内,主要由NLRP3、ASC和Caspase-1连接而成,能活化Caspase-1促进IL-1β和IL-18生成[12-14],其中IL-1β活化Th17细胞促进炎症反应[15],IL-18活化Th1细胞和NK细胞生成IFN-γ[16-17],二者协同作用对抗侵袭宿主的白色念珠菌。NLRP3炎症小体以依赖Caspase-1方式活化NF-κB信号途径[18],还能激活P38/MAPK通路和TAK1/JNK途径,诱导炎症反应及细胞焦亡[19-20]。
白色念珠菌细胞壁成份如β-葡聚糖、几丁质和甘露聚糖等是引发宿主固有免疫的诱导物。β-葡聚糖可以激活依赖于Dectin-1/syk通路的NLRP3炎症小体,诱导IL-1β的产生。宿主细胞依赖真菌葡聚糖诱导产生的ROS,通过NLRP3/ASC/Caspase-1轴诱导IL-1β分泌[21],破坏溶酶体导致了组织蛋白酶(比如组织蛋白酶 B和组织蛋白酶L等)泄露至胞质中,活化NLRP3[22];另外凝胶多糖(真菌的β-1,3-葡聚糖)还可以刺激宿主B淋巴细胞产生IgM抗体,而NLRP3基因缺陷宿主则不能产生相应抗体[23],这些提示NLRP3炎症小体在白色念珠菌细胞壁成份诱导的固有免疫甚至适应性免疫中均发挥重要的作用。进一步研究发现β-葡聚糖或白色念珠菌诱导的宿主细胞的炎症反应和细胞死亡,是由NLRP3-ASC-Caspase-1通路协同Dectin-1/CR3/Caspase-8通路共同完成,表明白色念珠菌感染时宿主可能通过多条信号通路针对白色念珠菌细胞壁成份即β-葡聚糖进行应答[24]。
白色念珠菌分泌型天冬氨酰蛋白酶(Sap),是真菌毒力的重要致病因子,可以分解宿主多种蛋白质。正是由于Sap利用分解蛋白质功能为白色念珠菌提供营养物质,并促进白色念珠菌粘附并侵入宿主细胞,造成宿主组织细胞损伤以及协助白色念珠菌从宿主细胞中逃逸[25]。Sap家族特别是Sap2和Sap6通过NLRP3-ASC-Caspase-1通路活化免疫细胞中的NLRP3炎症小体,继而产生IL-1β和IL-18,但经过Sap内化抑制剂处理后,IL-1β的产生明显减少,表明炎症小体的活化依赖网格蛋白诱导的Sap内化,以及K+内流和ROS的生成[26]。因此,在白色念珠菌感染过程中,Sap能够提高宿主抗真菌免疫力而不仅仅是协助真菌逃避免疫。Sap2和Sap6还能够通过旁路途径活化炎症小体。另外,Sap2和Sap6通过I型干扰素活化Caspase-11,后者协同Caspase-1共同诱导IL-1β和IL-18的分泌[27]。免疫细胞通过Caspase-11和Caspase-1之间的联系将炎症小体的经典活化与旁路活化结合起来,对抗白色念珠菌侵袭及其产生的Sap,这也说明宿主对抗白色念珠菌感染及其产生的致病因子应该是不同角度多方位进行应答的。
白色念珠菌具有酵母相和菌丝相两种存在形态,其中菌丝相是体现白色念珠菌致病能力的一个重要特征。研究表明白色念珠菌依赖NLRP3和Caspase-1诱导巨噬细胞焦亡,并且发现白色念珠菌菌株形态是影响焦亡以及IL-1β水平的重要因素[28]。灭活的酵母相白色念珠菌不会触发巨噬细胞焦亡,只能诱导低水平的IL-1β产生,而只有白色念珠菌非酵母相才能诱导免疫细胞焦亡,另外,选用在形态上能形成假菌丝而不能形成真菌丝的克柔念珠菌进行实验后,发现克柔念珠菌能够诱导巨噬细胞焦亡及活化NLRP3产生IL-1β,但产生的IL-1β水平低于白念珠菌实验组;选用形态上难以形成菌丝相的光滑念珠菌进行实验时基本上不触发焦亡及IL-1β的产生。另外,白色念珠菌UPC2突变株比未突变株更能诱导巨噬细胞焦亡,且能产生高水平的IL-1β;而白色念珠菌UPC2基因敲除株比未敲除株诱导巨噬细胞焦亡程度减轻,此时对白色念珠菌的菌丝并无明显影响,提示白色念珠菌所致焦亡也受到转录因子UPC2的调控[28]。综上所述,菌丝虽然是触发NLRP3诱导焦亡的重要因素,但却不是唯一的条件,因为转录因子UPC2也参与焦亡调控过程。
研究人员把形成菌丝的相关基因敲除后的白色念珠菌感染小鼠阴道,发现小鼠阴道冲洗液中中性粒细胞、钙结合蛋白与IL-1β水平,相比野生菌株均明显降低[29]。实验发现,白色念珠菌以NLRP3依赖方式诱导巨噬细胞分泌IL-1β,而不能形成菌丝的变异株则不能诱导巨噬细胞产生IL-1β;另外Nlrp3-/-小鼠感染白色念珠菌后,肝、脾、肾等脏器的真菌负荷明显高于对照野生型小鼠,说明Nlrp3-/-小鼠对白色念珠菌更加易感[30]。将白色念珠菌感染Nlrp3-/-小鼠的阴道,发现Nlrp3-/-小鼠阴道冲洗液中中性粒细胞减少,IL-1β、IL-6等炎性因子的分泌明显降低,此时宿主抗白色念珠菌的能力明显减弱[31]。这些实验证明菌丝相的白色念珠菌对于NLRP3炎性小体活化以及IL-1β的分泌相当重要,同时表明在清除白色念珠菌的固有免疫中NLRP3具有非常重要的作用。
白色念珠菌线粒体外膜/内质网膜上的4种蛋白质Mmm1/Mdm10/Mdm12/Mdm34形成的复合物即内质网(ER)-线粒体链(ERMES)对于维持线粒体形态以及菌体在宿主体内的生长非常重要[32]。为了准确观察巨噬细胞吞噬白色念珠菌后的炎症小体反应,用活细胞成像技术分析了白色念珠菌激活炎症小体时ERMES的作用。结果显示,Mmm1变异株及在NLRP3抑制剂MCC950作用下均使宿主NLRP3炎症小体活化程度降低和巨噬细胞焦亡程度减轻。ERMES活性丧失会破坏白色念珠菌线粒体形态,导致白色念珠菌菌丝相生长缓慢[32],提示ERMES与NLRP3炎症小体活化密切相关,也就是说白色念珠菌对NLRP3炎症小体的影响离不开自身线粒体形态的完整及菌丝的正常生长。另外对于平衡宿主炎症小体过度活化造成的免疫损伤与抗真菌免疫,还需要进一步研究。据ERMES突变体表型特征,且人体缺乏ERMES[33],表明ERMES是抗白色念珠菌治疗中充满前景的靶标。
2.2NLRP10在抗白色念珠菌感染中的作用NLRP10是唯一不具有C末端的亮氨酸重复序列(LRRs)结构域的NLR蛋白,主要分布在人和小鼠的肾脏组织、睾丸和结肠组织中。据推测NLRP10不识别PAMPs,可能在相关通路中仅发挥调控作用[34]。NLRP10通过其PYD结构域和ASC结合成复合物,再与Caspase-1活化NLRP3炎症小体;另外也与RIP2、TAK1和NEMO等蛋白质相互作用加强NOD1调节的免疫应答反应[35]。通过观察白色念珠菌感染Nlrp10-/-小鼠和WT小鼠实验后,发现Nlrp10-/-小鼠比WT小鼠更易感,且在体内并未影响NLRP3炎症小体的活化,但宿主体内细胞因子IFN-γ和IL-17的分泌量明显降低[36],显示Th1和Th17特异性应答反应明显减弱,表明NLRP10在宿主抗白色念珠菌感染的免疫过程中具有重要作用,但是参与宿主抗白色念珠菌感染的确切机制不清楚,有待于进一步的研究。
2.3NLRC4在抗白色念珠菌感染中的作用NLRC4也称IPAF、Card12以及CLAN,为胞浆内受体蛋白NLR家族成员[37],其C端为亮氨酸重复结构域(LRRs),具有识别PAMPs等配体的作用。中间为NACHT结构域,由NAIP、CIITA、HET-E和TP1组成,可介导NLRC4寡聚反应。N端为效应结构域,含三个Caspase募集结构域(CARD),可连接ASC和效应分子,转导下游信号。NLRC4发生多聚化,募集Caspase-1与ASC结合成炎症小体,诱导IL-1β产生,引起免疫应答[38]。研究发现正常小鼠口腔感染白色念珠菌后,口腔粘膜细胞中NLRC4、IL-1β、IL-6和IL-17的表达上升,以及舌背上皮中性粒细胞浸润程度增高,而感染白色念珠菌的Nlrc4-/-小鼠则降低,表明 NLRC4在宿主粘膜组织早期抗白色念珠菌感染中发挥作用[39]。但是白色念珠菌激活NLRC4炎性小体的具体机制并不清楚,有待今后的研究阐明。
2.4MDA5在抗白色念珠菌感染中的作用MDA5是RIG-I样受体家族的重要成员,有诱导细胞终端分化作用。N端包括2个相同的CARD,中间为DExD/H解旋酶区,C端是没有活性的抑制域(RD)。MDA5识别细小RNA病毒,依赖CARD结构域与接头蛋白IPS-1相互作用,活化IRF-3/IRF-7,激活NF-κB,促进IFN-β的表达,从而引起宿主先天免疫反应[40]。
采用白色念珠菌的酵母相或菌丝相刺激来源于Mda5-/-小鼠的脾细胞后,结果显示细胞不能产生IFN-β,细胞因子IL-6和IL-10表达水平也较低,同时表明在宿主防御白色念珠菌感染的过程中MDA 5起着重要的作用[41]。Ⅰ型干扰素在抗真菌感染中起重要作用,MDA5可能作为一种受体部分参与了白色念珠菌感染时Ⅰ型干扰素的表达,MDA5突变导致表达或活性的增加,可能会增加IFN的产生[42-43]。I型干扰素的异常产生反过来又会引起免疫反应的不平衡。IfnarI-/-小鼠对系统性念珠菌感染的抵抗力更强[44],Mda5-/-小鼠对播散性念珠菌病更有抵抗力[41],提示MDA5在抗白色念珠菌感染过程中起负调控作用。目前,除了RNAs是MDA5的配体,白色念珠菌感染后特异识别并激活MDA5是否还有其它配体及确切机制尚不清楚,有待进一步研究。
3 结论与展望
胞内模式识别受体通过识别白色念珠菌的PAMPs,活化宿主固有免疫,继而启动适应性免疫,启动机体抗白色念珠菌感染的免疫反应(图1)。但是胞内模式受体识别配体、活化下游信号通路以及胞内模式受体间的确切机制仍然不是很清楚,还需深入探究。以NLRP3、ASC和Caspase-1为核心的NLRP3炎症小体,参与了人类众多疾病的发生发展[45-47]。小分子抑制剂(CY-09)能够抑制NLRP3炎症小体的活化和IL-1β的产生[48],在NLRP3相关的炎症型疾病动物模型中,曲尼斯特能够治疗NLRP3驱动的相关炎症性疾病[49],提示胞内模式识别受体可作为一种潜在的药物靶标。目前抗真菌感染的药物大多具有毒性,在临床应用方面这些药物又有其局限性,且随着抗真菌药物的大量应用,不断出现真菌耐药性,抗真菌治疗也变得举步维艰。胞内模式识别受体及其信号通路相关分子可作为潜在的药物靶点,为抗白色念珠菌感染的药物开发和真菌病的治疗提供新的思路和路径。
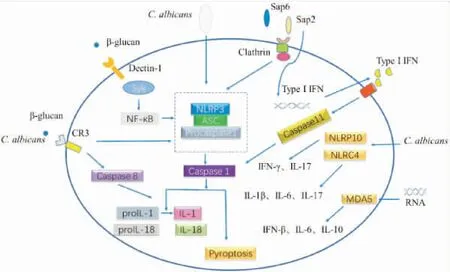
图1 模式识别受体在抗白色念珠菌感染中的作用
