异养硝化-好氧反硝化菌富集驯化过程中微生物种群演变特征——典型城市景观水系
2019-11-28周石磊张艺冉杨文丽黄廷林李再兴崔建升周子振
周石磊,张艺冉,孙 悦,杨文丽,黄廷林,李再兴,罗 晓,崔建升,周子振,李 扬
异养硝化-好氧反硝化菌富集驯化过程中微生物种群演变特征——典型城市景观水系
周石磊1*,张艺冉1,孙 悦1,杨文丽1,黄廷林2,李再兴1,罗 晓1,崔建升1,周子振3,李 扬3
(1.河北科技大学环境科学与工程学院,河北省污染防治生物技术实验室,河北 石家庄 050018;2.西安建筑科技大学环境与市政工程学院,西北水资源与环境生态教育部重点实验室,陕西 西安 710055;3.中原工学院能源与环境学院,河南 郑州 450007)
为了研究不同异养硝化-好氧反硝化菌富集驯化过程中水体微生物群落的演变,利用Miseq 高通量测序法对景观水系沉积物富集驯化样本的微生物信息进行统计,对其微生物群落的多样性以及多样性进行分析,同时基于微生物属的信息进行了微生物网络分析.结果显示,两种类型培养基在富集驯化完成后氮素得到有效去除,脱氮效果明显;富集驯化过程中的OUT主要属于7个,分别是变形菌门(Protebacterice)、拟杆菌门(Bacteroidetes)、绿弯菌门(Chloroflexi)、厚壁菌门(Firmicutes)、放线菌门(Actinobacteria)、蓝藻门(Cyanobacteria)、酸杆菌门(Acidobacteria),与此同时,富集驯化过程中有关氮循环的细菌有上升的变化过程;主成分分析(PCA),非度量多维尺度分析(NMDS)以及主坐标分析(PCoA)表明异养硝化-好氧反硝化菌富集驯化过程中不同温度压力下的细菌群落组成存在明显差异,而培养基的类别带来的影响相对较小;网络分析显示模块核心和网络核心均为低丰度的稀有物种;膨胀因子分析(VIF)和冗余分析(RDA)得出温度、氨氮和硝酸盐氮是影响群落结构演变的关键环境因子.综上可知,Miseq高通量测序研究异养硝化-好氧反硝化菌富集驯化过程中微生物种群演变可行,为实现微生物菌剂“定向-精准-高效”的筛选提供技术支撑.
异养硝化-好氧反硝化;景观水系;Miseq测序;生物信息分析;微生物群落
好氧反硝化菌[1]是一类在有氧条件下,利用周质硝酸盐还原酶进行反硝化作用的脱氮菌,并且大多数菌同时具有异养硝化的能力.它的发现打破了反硝化只能在厌氧缺氧条件下进行的传统反硝化的观念,为新型生物脱氮提供了新思路.
近年来,相关研究通过从土壤[2]、活性污泥[3]、污水[4-5]、河流[6]、湖泊[7-8]、水库[9-10]等系统,分离了大量高效的好氧反硝化菌.前期文献报道主要集中于通过施加一定选择压(间歇曝气、盐度或者温度)或采用特定的选择性培养基驯化富集.比如,赵惊鸿等[11]通过采取在好氧反硝化培养基连续培养中逐步升温的方式,分离得到耐高温好氧反硝化菌JH8;成钰等[12]将通过在好氧反硝化培养液中添加氯化钠的方式,分离得到耐盐的异养硝化-好氧反硝化芽孢杆菌SLWX2;周培等[13]通过特定方法完成耐受重金属好氧反硝化菌株的筛选;Carter等[14]利用周质硝酸盐还原酶特定的生化特性和活性位点,筛选出多种好氧反硝化菌;Kong等[15]利用氰化钾作为抑制剂,快速筛选出好氧反硝化菌.然而,关于在好氧反硝化菌富集驯化过程中微生物群落的演变过程鲜有报道.不同富集驯化方法下微生物群落演变规律直接影响高效好氧反硝化菌的定向筛选和特性研究,特别是对高效菌(混合菌群)的潜在功能预测以及菌剂的固定化提供必要依据.
石家庄中心城区水系作为省会的核心水系,保持健康的生态功能为城市的经济发展提供了重要的生态保障.由于水系氮素超标严重,且主要为氨氮和硝酸盐氮.完成黑臭水体治理,首先要恢复水体的自然复氧功能,使水体呈现好氧状态.因此,如何在好氧环境下实现“定向-精准-高效”的氮素削减是水体自我修复功能恢复的关键,同时也是一个亟待解决的科学问题.本文选取石家庄中心城区典型区域的沉积物样品,采用不同的异养硝化-好氧反硝化培养基进行常温和低温条件下异养硝化-好氧反硝化菌的富集驯化;与此同时研究富集驯化过程中的微生物群落演变特征,考察关键异养硝化-好氧反硝化物种,为进一步的高效菌剂定向筛选提供必要的技术支持.
1 材料与方法
1.1 实验装置
实验装置为2L的烧杯,取石家庄市中心城区世纪公园(SJ)(38°11′40″N;114°32′16″E)和民心河裕翔街(YX)(37°58′41″N;114°31′45″E)的沉积物进行好氧反硝化菌的富集驯化.通过恒温培养箱和充氧泵的间歇曝气来控制系统的温度和溶解氧,实验的温度设为室温20℃和低温10℃.每个2L烧杯中装有200mL的沉积物和800mL的培养基,其中沉积物样品用超纯水进行了洗脱处理.通过选择2种类型异养硝化-好氧反硝化培养基来富集驯化好氧反硝化菌.具体富集驯化过程为,首次为100%培养基,进行培养;3d后更换培养基为100%的培养基继续培养;3d后更换培养基为80%的培养基培养;3d后更换为70%的培养基培养;3d后更换60%的培养基培养;3d后更换50%的灭菌富集培养基培养.每次更换期间测定各个系统水样中硝酸盐氮、亚硝酸盐氮、氨氮和总氮来反应异养硝化-好氧反硝化菌的富集驯化效果.
1.2 异养硝化-好氧反硝化培养基
异养硝化-好氧反硝化(类型Ⅰ)富集培养基[16](g/L):CH3COONa 0.1;NaNO30.01;NH4Cl 0.0063;K2HPO4·3H2O 0.02;CaCl20.01;MgCl2·6H2O 0.01;蒸馏水1L;pH 7.0 ~7.5.
异养硝化-常规反硝化(类型Ⅱ)富集培养基[17](g/L):Na2HPO4·7H2O 7.9;KH2PO41.5;MgSO4·7H2O 0.1;丁二酸钠(琥珀酸钠)4.7;KNO31.0;微量元素2mL/L;pH值 7.0~7.5.微量元素(g/L):EDTA 50; ZnSO42.2;CaCl25.5;MnCl2·4H2O 5.1;FeSO4·7H2O 5.0;钼酸盐1.1;CuSO4·5H2O 1.6;CoCl2·6H2O 1.6;蒸馏水1L; pH 7.0~7.5.
本研究调整类型Ⅱ的浓度(稀释30倍),使其与类型Ⅰ的氮素水平相一致,以便城市景观水系的氮素污染控制.
1.3 微生物多样性分析
1.3.1 DNA提取与PCR扩增 选取初始的世纪公园和民心河的新鲜沉积物样品以及富集驯化结束后的沉积物样品,通过土壤DNA试剂盒(美国MP公司FastDNA SPINTMkit)并参照说明书提取沉积物的总DNA,采用琼脂糖凝胶电泳法和分光光度计完成DNA完整性、纯度与浓度检测.沉积物在-80℃下保存,以备高通量测序.
1.3.2 微生物16S测序 将提取的沉积物总DNA样本利用引物338F(ACTCCTACGGGAGGCAGC-AG)和806R(GGACTACHVGGGTWTCTAAT)对沉积物的16S rRNA基因V4-V5区进行PCR扩增,进而分析异养硝化-好氧反硝化菌富集驯化过程中微生物群落的演变过程.20μL PCR反应体系为:5×FastPfu Buffer 4μL, 2.5mmol/L dNTPs 2μL, Forward Primer (5μmol/L) 0.8μL, Reverse Primer(5μmol/L) 0.8μL, FastPfu Polymerase 0.4μL, BSA 0.2μL, Template DNA 10ng, ddH2O 11.8mL.反应程序为:初始变性温度95℃ 3.0min; 95℃ 30s, 55℃ 30s, 72℃ 45s,循环30次; 72℃ 10min读板.将PCR扩增的产物委托上海美吉生物科技有限公司利用Miseq进行高通量测序.
1.3.3 生物信息学分析 对测序结果进行数据质控,依据相似度97%水平划分OUT,并按照最小样本序列进行抽平处理.利用R语言分析微生物群落的多样性以及多样性分析(http://www.r-project.org/).基于R语言的vegan包完成主成分分析(PCA),非度量多维尺度分析(NMDS)以及主坐标分析(PCoA),进而分析不同培养基下异养硝化-好氧反硝化菌富集驯化过程中的微生物群落组成差异.基于方差膨胀因子(VIF)分析以及蒙特卡罗检验筛选关键影响因子[18],并通过冗余分析(RDA)得到环境因子与微生物群落之间的相关性[19]. 利用Cytoscape软件进行微生物群落的网络分析[20],筛选出关键物种[21],进而分析不同选择压下富集驯化得到的异养硝化-好氧反硝化菌的物种信息.
1.4 水质分析方法
水质指标硝酸盐氮采用紫外分光光度法,亚硝酸盐氮为N-(1-奈基)-乙二胺光度法,氨氮为纳氏试剂比色法,总氮为过硫酸钾氧化-紫外分光光度法.硝酸盐氮、亚硝酸盐氮和氨氮的水样经0.45μm醋酸纤维滤膜过滤.
1.5 数据分析方法
绘图和数据统计分析软件为R和Origin.其中值<0.05表示存在显著差异,0.001<值<0.01表示存在极显著差异,值<0.001表示存在极其显著差异.参照文献[22],网络分析中对网络中节点进行如下分类:模块核心(Z³2.5,P£0.62),网络核心(Z³0.5,P>0.62),外围节点(Z<2.5,P£0.62),连接点(Z<2.5,P>0.62).
2 结果与分析
2.1 源水脱氮效果分析
如图1所示,两种类型培养基在从100%培养基到50%培养基的富集驯化过程中,都表现出很强的好氧反硝化脱氮能力.类型Ⅰ培养基下,YX采样点在低温的反硝化能力要强于常温,比如在50%的培养基条件下,低温环境3d的YX的硝酸盐氮从5.44mg/L下降到0.30mg/L,而常温条件下硝酸盐氮从5.44mg/L下降到1.14mg/L;低温条件下SJ的硝酸盐氮从1.30mg/L下降到0.17mg/L,而常温条件下硝酸盐氮从1.30mg/L下降到0.34mg/L;YX和SJ低温与常温反硝化能力的不同,可能与本底微生物群落组成有关,具体原因还需进一步分析.类型Ⅱ的培养基低温和常温的好氧反硝化脱氮能力差异不大,YX的硝酸盐氮去除率82.65%~84.16%;SJ的硝酸盐氮去除率89.48%~90.77%.然而,在异养硝化-好氧反硝化菌富集驯化过程中,两种培养基的氨氮并没有表现出明显的去除效果.其中,在结束富集驯化时常温下的氨氮表现出更好的去除效果.比如,类型Ⅰ的培养基YX的氨氮从3.55mg/L下降到0.52mg/L,SJ的氨氮从0.75mg/L下降到0.69mg/L;类型Ⅱ的培养基YX的氨氮从2.53mg/L下降到1.39mg/L,SJ的氨氮从0.05mg/L上升到1.47mg/L.氨氮去除不明显的原因可能与沉积物的释放有关.
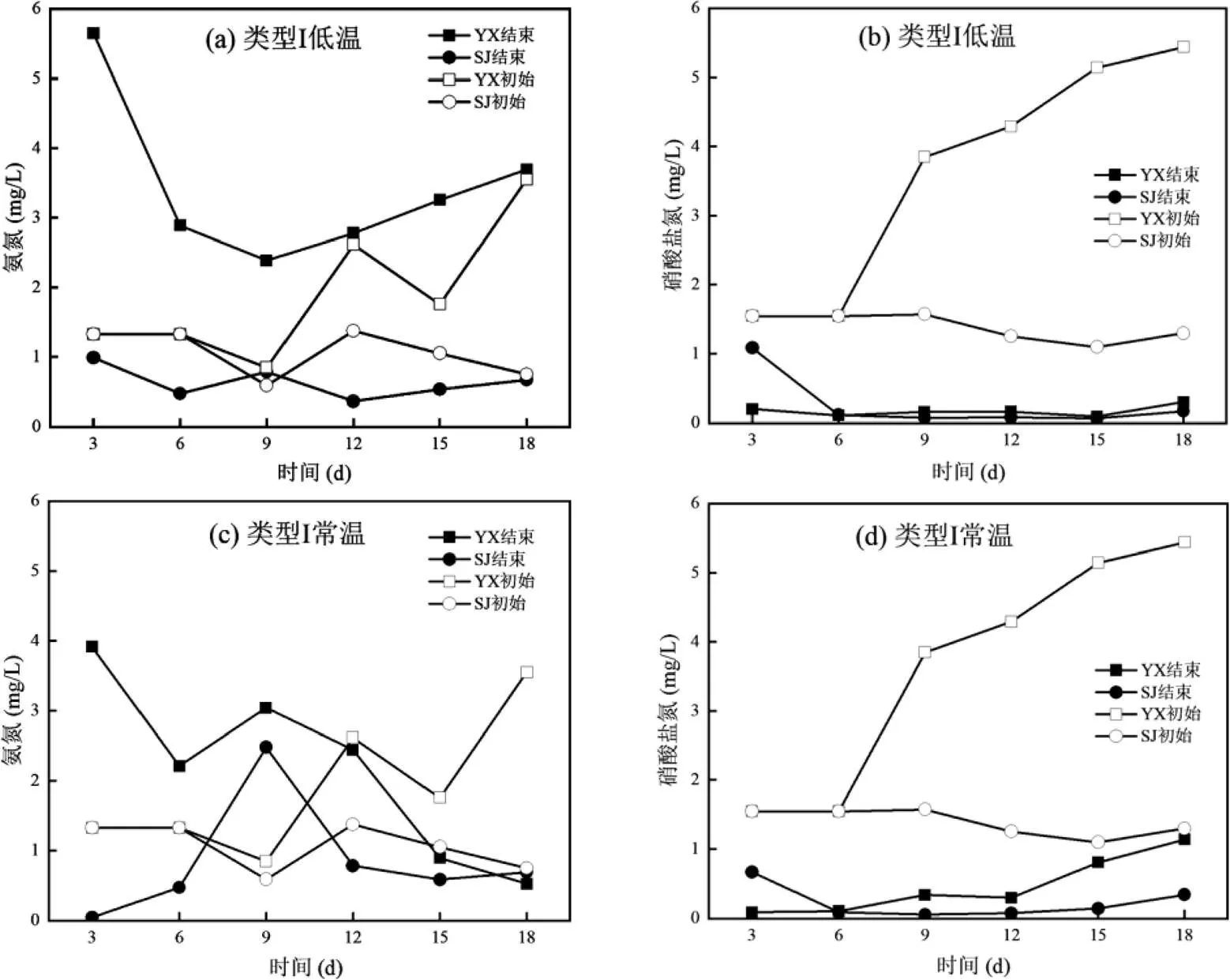
图1 异养硝化-好氧反硝化菌富集驯化过程中氮素变化特征
Fig.1 The changes of nitrogen concentrations during the enrichment and domestication process of heterotrophic nitrification-aerobic denitrification bacteria
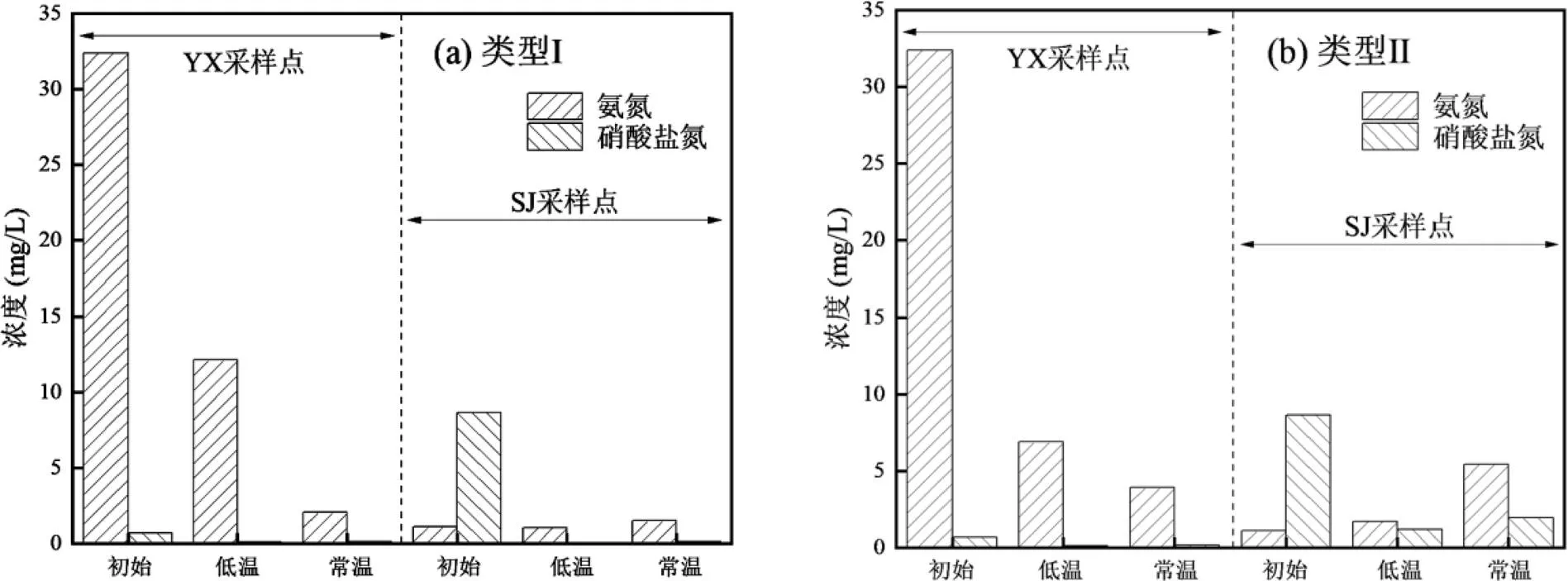
图2 异养硝化-好氧反硝化菌富集驯化过程中间隙水氮素变化特征
关于富集驯化过程中各个系统的沉积物间隙水中氮素的变化情况如图2所示.YX采样点的氨氮呈现明显的降低,类型Ⅰ的培养基从初始的32.39mg/L下降到12.14(低温)和2.07mg/L(常温);类型Ⅱ的培养基从初始的32.39mg/L下降到6.90(低温)和3.93mg/L(常温).SJ采样点的硝酸盐氮呈现明显的降低,类型Ⅰ的培养基从初始的8.67mg/L下降到0.05 (低温)和0.15mg/L(常温);类型Ⅱ的培养基从初始的8.67mg/L下降到1.21(低温)和1.95mg/L(常温). YX采样点表现出更高的氨氮去除效果,SJ采样点表现出更高的硝酸盐氮去除效果.
2.2 微生物α多样性分析
通过对两种培养基异养硝化-好氧反硝化菌富集驯化样本16S rRNA基因进行测序,总共获得了34881条有效序列,序列平均长度440bp.通过计算微生物多样性指数考察不同类型培养基富集驯化异养硝化-好氧反硝化菌实验过程中细菌微生物群落的物种丰度和物种多样性.
如表1所示,Ace和Chao指数反映微生物群落的丰富度[23],两种类型培养基下的微生物丰富度都有明显的增加,Ace指数从2120.78增加到2319.69, Chao指数从2146.76增加到2344.23; Coverage反映微生物群落的覆盖度[24],在富集驯化过程中维持在0.9887~0.9925之间,表明测序可以很好的覆盖物种信息;Shannon和Simpson指数反映微生物群落的多样性[25],Shannon指数大多数呈现增加,然而Simpson指数大多数为降低,微生物多样性变化的复杂原因还需进一步分析.
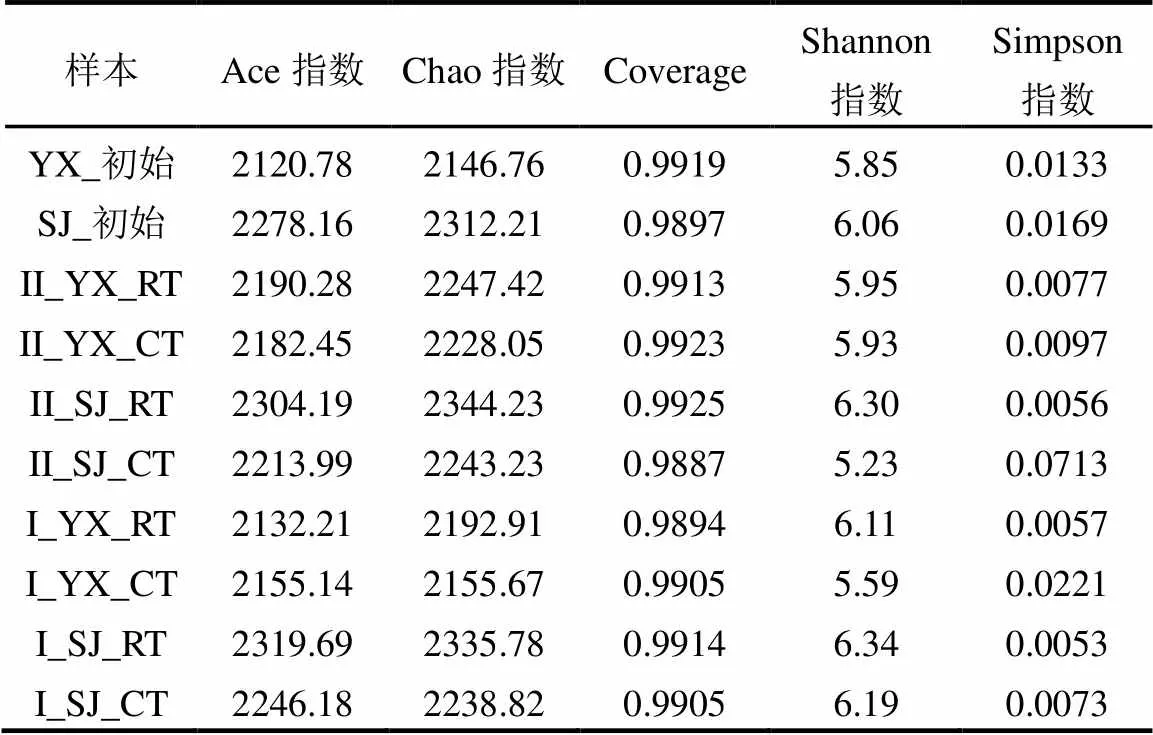
表1 异养硝化-好氧反硝化菌富集驯化过程中生物多样性指数
2.3 细菌群落组成及β多样性分析
将异养硝化-好氧反硝化菌富集驯化过程中的样本进行数据库比对,同时分析在各个水平上的菌群结构.结果显示属于52个门类866个属3081个OTU,具体结果如图3所示.
两种类型培养基在低温和常温条件下富集驯化的OUT主要属于7个门类如图3(a),分别是变形菌门 (Protebacterice)、拟杆菌门(Bacteroidetes)、绿弯菌门(Chloroflexi)、厚壁菌门(Firmicutes)、放线菌门 (Actinobacteria)、蓝藻门(Cyanobacteria)、酸杆菌门(Acidobacteria)和其他少数细菌门类,其中主要的细菌门类为变形菌门.特别是,变形菌门在碳源和氮素代谢过程中扮演重要的角色[26];拟杆菌门主要参与硝化过程[27];绿弯菌门能够促进水中植物残留物的降解过程[28].其中变形菌门占比34.89%~ 68.06%,拟杆菌门占比7.29%~15.67%,绿弯菌门占比6.59%~19.39%.
在变形菌门中(图3(b)),各个异养硝化-好氧反硝化菌富集驯化过程中共包含Alphaproteobacteria, Betaproteobacteria,Gammaproteobacteria,Deltaproteobacteria和Epsilonproteobacteria 5种.其中, Betaproteobacteria是最大的纲,而且Betaproteobacteria因其具有氮素去除的降解特征[29],广泛分布污废水的处理系统[30].本实验中,YX从初始的13.83%上升到23.22%;SJ从初始的14.62%上升到44.87%,表明在异养硝化-好氧反硝化菌富集驯化过程中,脱氮微生物得到明显增加.
与此同时,两种培养基在富集驯化过程中微生物种群存在共有的属种,同时也存在差异.现将富集驯化过程中丰度前50的属如图3(c)所示,其中很多涉及氮素循环.比如,在培养基Ⅰ中,YX采样点中的,,,以及得到增加,分别从6.31%, 1.39%, 0.63%, 0.14%, 0.81% 上升到10.56%, 3.56%, 1.93%, 10.46%, 1.81%; SJ采样点中的,,,以及也得到增加,分别从2.95%, 1.98%, 1.79%, 0.24%, 0.16%上升到4.64%, 3.34%, 2.34%, 1.06%, 1.21%.在培养基Ⅱ中,YX采样点中的,,和,得到增加,分别从1.39%, 0.14%, 0.63%, 1.11%上升到3.17%, 3.75%, 1.85%, 1.56%;SJ采样点中的,,,和得到增加,分别从0.24%, 0.74%, 0.01%, 0.16%上升到1.47%, 1.34%, 24.60%, 1.06%.其中,作为绿弯菌门的代表类群,属于有机物降解的一类微生物[31];和促进氮素和有机物的去除[32-33],并且作为异养硝化-好氧反硝化菌主要属种[34];和是主要的反硝化菌[35-36],反硝化菌在废水硝酸盐氮去除过程中得到增加[37];作为代表性异养硝化-好氧反硝化菌分离于废水[38]、沉积物[39]和污泥[40]中;与[41]在系统发育上接近,也是重要的反硝化菌[42].

图3 异养硝化-好氧反硝化菌富集驯化过程中种群变化特征
与此同时,基于OTU (97%相似性)水平,对两种培养基类型的异养硝化-好氧反硝化菌富集过程的种群演变进行了β-多样性的分析,具体包括:主成分分析(PCA),非度量多维尺度分析(NMDS)以及主坐标分析(PCoA),来反映不同培养基以及不同温度下富集驯化的菌种差异性(图4).PCA分析得出PC1+PC2达到58.35%,图中样本间的组成越相似,反映在PCA图中的距离越近(图4(a)).PCA图中两类培养基分布在PCA1轴的正负两侧,同一温度条件下的样本聚集相对紧密,低温和常温样本间分布较分散,显示2个采样点在富集驯化过程中种群呈现差异性.NMDS中当stress<0.05时,则具有很好的代表性[43],本文分析显示不同采样点在2种培养基中,在富集驯化过程中物种呈现显著差异,同一温度的差异较小(图4(b)).通过基于 Bray–Curtis距离的PCoA分析研究不同培养基类型富集驯化异养硝化-好氧反硝化菌的群落组成(图4(c)).结果表明,不同温度压力下的细菌群落组成存在明显差异,而培养基的类别带来的影响相对较小,跟PCA、NMDS分析以及物种群落组成的结果相一致.
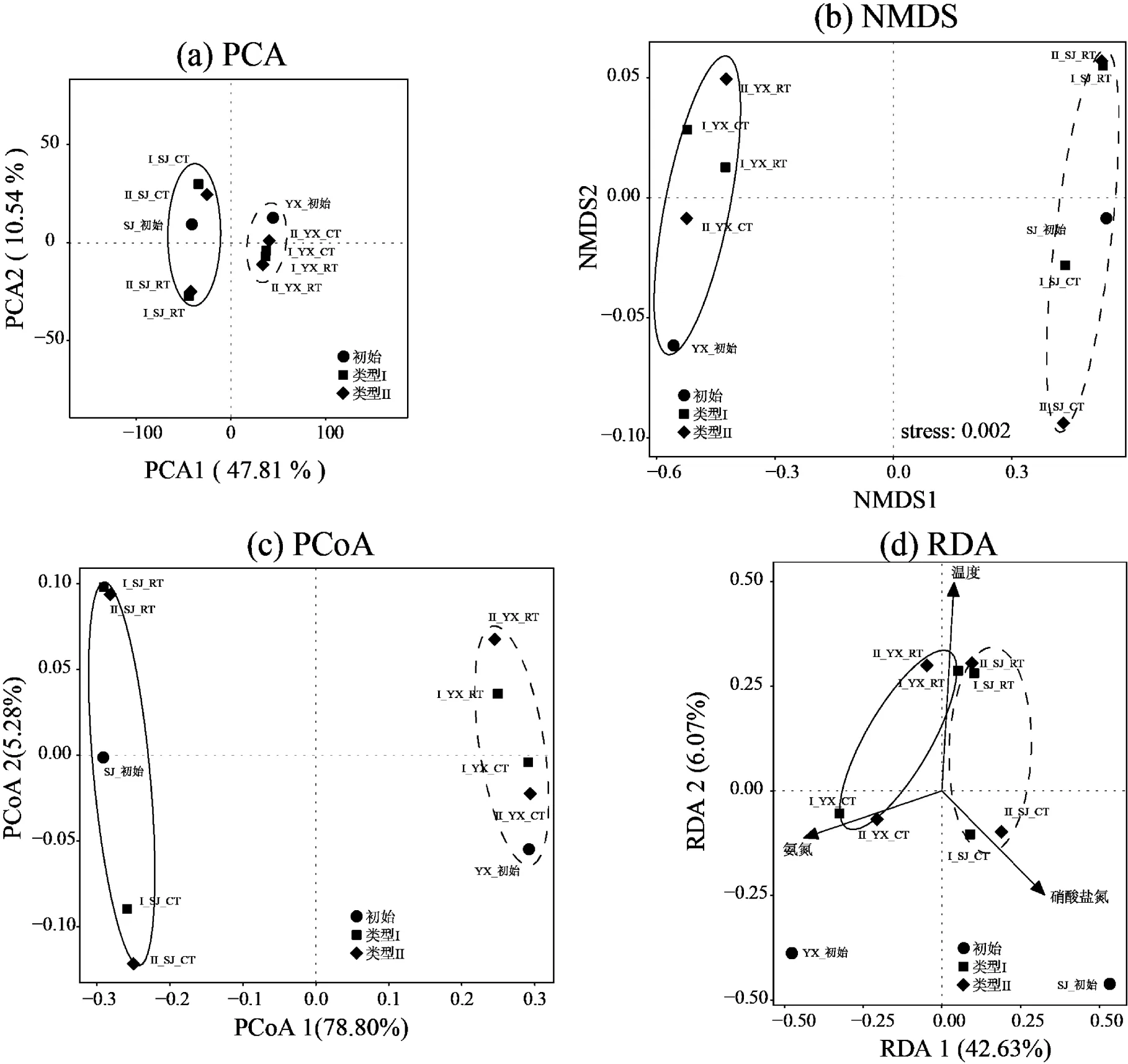
图4 异养硝化-好氧反硝化菌富集驯化过程中种群差异分析
2.4 微生物网络分析
基于异养硝化-好氧反硝化菌富集驯化过程中的微生物群落的属,利用Cytoscape软件构建微生物的互作网络.
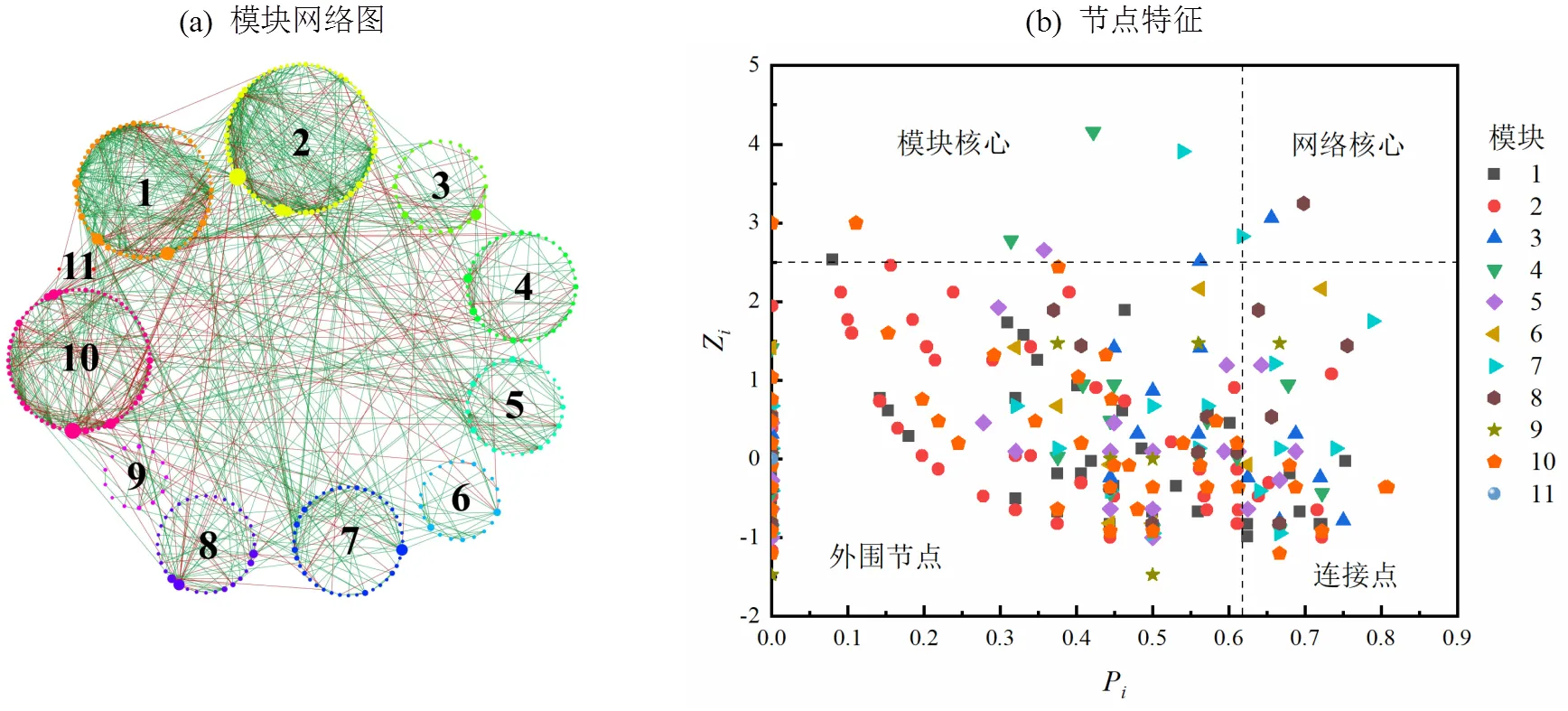
图5 异养硝化-好氧反硝化菌富集驯化过程中微生物网络及节点特征
图5(a)中展示为斯皮尔曼相关系数>0.99的节点,节点大小为中介中心性,线的颜色绿色表示正相关,红色表示负相关.网络分析共得到412个节点,1327条边;划分成11个模块,模块1~11的占比分别为15.05%, 18.20%, 6.80%, 9.71%, 7.52%, 5.10%, 9.71%, 7.77%, 3.16%, 16.50%, 0.48%.网络中节点正相关占比74.00%,负相关占比26.00%,表明物种间的关系呈现共生为主.Roger等[44]定义参数Z来衡量一个点在所在模块中的作用,值越高说明在模块中的作用越大;定义P来衡量一个点参与其他模块的程度,值越高说明与其他模块的联系越密切.有关节点的分类如图5(b)所示,网络分析依据各节点Z值和P值将所有节点划分为模块核心,网络核心,外围节点和连接点四类.本网络中模块核心包括14个物种,网络核心包括2个物种.模块核心和网络核心大多属于低丰度的稀有物种,其中,能利用各种蛋白质进行生长[45],是一种降解有机物的关键物种[46-47];是污水处理厂的一个活性反硝化菌[48],说明稀有种在异养硝化-好氧反硝化菌富集驯化过程中对群落构建发挥着不可替代的作用.
2.5 环境因子与微生物群落的关系
基于VIF分析和蒙特卡罗检验,得到关键环境因子温度(VIF=1.63<10),氨氮(VIF=1.34<10)和硝酸盐氮(VIF=1.55<10).RDA分析显示RDA1和RDA2共解释了总体变化的48.7%,其中RDA1占主体解释了42.63%(图4(d)).其中,温度与RDA1相关性达到-0.14(=0.001<0.05),与RDA2相关性达到-0.99(= 0.001<0.05);氨氮与RDA1相关性达到-0.89(= 0.03<0.05),与RDA2相关性达到-0.45(=0.003< 0.05);硝酸盐氮与RDA1相关性达到0.66(=0.03< 0.05),与RDA2相关性达到-0.75(=0.03<0.05).综上,温度决定了两种培养基不同采样点的不同温度物种分布,常温的富集驯化过程菌群分布在RDA2的正向,低温的富集驯化过程菌群分布在RDA2的正向;氨氮和硝酸盐氮分别与YX和SJ采样点的富集驯化过程中菌群成正相关.
3 结论
3.1 类型Ⅰ培养基下,YX采样点在低温的反硝化能力要强于常温;类型Ⅱ的培养基下2采样点在低温和常温的反硝化脱氮能力差异不大.
3.2 两种类型培养基在低温和常温条件下富集驯化的OUT主要属于变形菌门、拟杆菌门、绿弯菌门、厚壁菌门、放线菌门、蓝藻门、酸杆菌门.
3.3 不同温度压力下的细菌群落组成存在明显差异, 而培养基的类别影响相对较小;温度、氨氮以及硝酸盐氮是影响群落结构的关键环境因子.微生物网络中物种大多呈现共生关系, 关键节点显示物种大多为稀有物种.
[1] Gao H, Schreiber F, Collins G, et al. Aerobic denitrification in permeable Wadden Sea sediments [J]. The ISME journal, 2010,4(3):417-426.
[2] He T, Li Z, Sun Q, et al. Heterotrophic nitrification and aerobic denitrification byY-11without nitrite accumulation during nitrogen conversion [J]. Bioresource Technology, 2016,200:493-499.
[3] Lu Z, Gan L, Lin J, et al. Aerobic denitrification bysp. YF1in the presence of Cu(II) [J]. Science of the Total Environment, 2019,658:80-86.
[4] Yang J R, Wang Y, Chen H, et al. Ammonium removal characteristics of an acid-resistant bacteriumsp. JR1from pharmaceutical wastewater capable of heterotrophic nitrification- aerobic denitrification [J]. Bioresource Technology, 2019,274:56-64.
[5] Zhao B, Cheng D Y, Tan P, et al. Characterization of an aerobic denitrifierstrain XL-2 to achieve efficient nitrate removal [J]. Bioresource Technology, 2018,250:564-573.
[6] Jin P, Chen Y, Yao R, et al. New insight into the nitrogen metabolism of simultaneous heterotrophic nitrification-aerobic denitrification bacterium in mRNA expression [J]. J. Hazardous Materials, 2019,371:295-303.
[7] Su J F, Shi J X, Ma F. Aerobic denitrification and biomineralization by a novel heterotrophic bacterium,sp. H36 [J]. Marine Pollution Bulletin, 2017,116(1):209-215.
[8] Li C, Yang J, Wang X, et al. Removal of nitrogen by heterotrophic nitrification–aerobic denitrification of a phosphate accumulating bacteriumYG-24 [J]. Bioresource Technology, 2015,182:18-25.
[9] Huang T L, Zhou S L, Zhang H H, et al. Nitrogen removal characteristics of a newly isolated indigenous aerobic denitrifier from oligotrophic drinking water reservoir,sp. N299 [J]. International Journal of Molecular Sciences, 2015,16(5):10038-10060.
[10] 周石磊,黄廷林,白士远,等.贫营养好氧反硝化菌的分离鉴定及其脱氮特性 [J]. 中国环境科学, 2016,36(1):238-248. Zhou S L, Huang T L, Bai S Y, et al. Isolation, identification, and nitrogen removal characteristics of oligotrophic aerobic denitrifiers [J]. China Environmental Science, 2016,36(1):238-248.
[11] 赵惊鸿,黄少斌.一株耐高温好氧反硝化菌的筛选及特性研究 [J]. 环境科学与技术, 2015,38(1):6-10+67. Zhao J H, Haung S B. Isolation and characteristics of a thermophilic aerobic denitrifier [J]. Environmental Science & Technology, 2015, 38(1):6-10+67.
[12] 成 钰,李秋芬,费聿涛,等.海水异养硝化-好氧反硝化芽孢杆菌SLWX_2的筛选及脱氮特性 [J]. 环境科学, 2016,37(7):2681-2688. Cheng Y, Li Q F, Fei Y T, et al. Screening and nitrogen removing characteristics of heterotrophic nitrification-aerobic denitrification bacteria SLWX2from sea water [J]. Environmental Science, 2016, 37(7):2681-2688.
[13] 周 培,张 丹,支月娥,等.耐受重金属的好氧反硝化菌株及其应用, CN104152377A [P/OL]. 2014-11-19].
[14] Carter J P, Hsaio Y, Spiro S, et al. Soil and sediment bacteria capable of aerobic nitrate respiration [J]. Applied and Environmental Microbiology, 1995,61(8):2852-2858.
[15] Kong Q X, Wang X W, Jin M, et al. Development and application of a novel and effective screening method for aerobic denitrifying bacteria [J]. FEMS Microbiology Letters, 2006,260(2):150-155.
[16] 黄廷林,白士远,张海涵,等.一株贫营养异养硝化-好氧反硝化细菌的分离鉴定及脱氮特性 [J]. 环境工程学报, 2015,9(12):5665-5671. Huang T L, Bai S Y, Zhang H H, et al. Identification and denitrification characteristics of an oligotrophic heterotrophic nitrification and aerobic denitrification bacteria [J]. Chinese Journal of Environmental Engineering, 2015,9(12):5665-5671.
[17] Zhu L, Ding W, Feng L J, et al. Isolation of aerobic denitrifiers and characterization for their potential application in the bioremediation of oligotrophic ecosystem [J]. Bioresource Technology, 2012,108:1-7.
[18] Joshi D R, Zhang Y, Gao Y, et al. Biotransformation of nitrogen-and sulfur-containing pollutants during coking wastewater treatment: Correspondence of performance to microbial community functional structure [J]. Water Research, 2017,121:338-348.
[19] Kernan M R, Helliwell R C. Partitioning the variation within the acid neutralizing capacity of surface waters in Scotland in relation to land cover, soil and atmospheric depositional factors [J]. Science of The Total Environment, 2001,265(1):39-49.
[20] Zhang K, Gu J, Wang X, et al. Variations in the denitrifying microbial community and functional genes during mesophilic and thermophilic anaerobic digestion of cattle manure [J]. Science of The Total Environment, 2018,634:501-508.
[21] Banerjee S, Baah-acheamfour M, Carlyle C N, et al. Determinants of bacterial communities in Canadian agroforestry systems [J]. Environmental Microbiology, 2016,18(6):1805-1816.
[22] Jizhong Z. Phylogenetic molecular ecological network of soil microbial communities in response to elevated CO2[J]. mBio, 2011, 2(4):e00122-11.
[23] Rogers M B, Firek B, Shi M, et al. Disruption of the microbiota across multiple body sites in critically ill children [J]. Microbiome, 2016, 4(1):66.
[24] Xin X, He J, Wang Y, et al. Role of aeration intensity on performance and microbial community profiles in a sequencing batch reaction kettle (SBRK) for wastewater nutrients rapid removal [J]. Bioresource Technology, 2016,201:140-147.
[25] Chen B, Teh B S, Sun C, et al. Biodiversity and activity of the gut microbiota across the life history of the insect herbivore Spodoptera littoralis [J]. Scientific Reports, 2016,6:29505.
[26] Hou L, Zhou Q, Wu Q, et al. Spatiotemporal changes in bacterial community and microbial activity in a full-scale drinking water treatment plant [J]. Science of the Total Environment, 2018,625:449-459.
[27] Wu D, Zhang Z, Yu Z, et al. Optimization of F/M ratio for stability of aerobic granular process via quantitative sludge discharge [J]. Bioresoure Technology, 2018,252:150-156.
[28] Kielak A M, Barreto C C, Kowalchuk G A, et al. The ecology of acidobacteria: Moving beyond Genes and Genomes [J]. Frontiers in Microbiology, 2016,7:744.
[29] Jie G, Yu D, Ying L, et al. Long- and short-chain AHLs affect AOA and AOB microbial community composition and ammonia oxidation rate in activated sludge [J]. J. Environmental Sciences-China, 2018,78:53-62.
[30] Figuerola E L M, Leonardo E. Bacterial taxa abundance pattern in an industrial wastewater treatment system determined by the full rRNA cycle approach [J]. Environmental Microbiology, 2010,9(7):1780-1789.
[31] Li Z, Kechen X, Yongzhen P. Composition characterization and transformation mechanism of refractory dissolved organic matter from an ANAMMOX reactor fed with mature landfill leachate [J]. Bioresource Technology, 2018,250:413-421.
[32] Zhang X, Fu W, Yin Y, et al. Adsorption-reduction removal of Cr(VI) by tobacco petiole pyrolytic biochar: Batch experiment, kinetic and mechanism studies [J]. Bioresoure Technology, 2018,268:149-157.
[33] Kong X X, Jiang J L, Qiao B, et al. The biodegradation of cefuroxime, cefotaxime and cefpirome by the synthetic consortium with probiotic Bacillus clausii and investigation of their potential biodegradation pathways [J]. Science of the Total Environment, 2019,651:271-280.
[34] Chen G, Huang J, Fang Y, et al. Microbial community succession and pollutants removal of a novel carriers enhanced duckweed treatment system for rural wastewater in Dianchi Lake basin [J]. Bioresoure Technology, 2019,276:8-17.
[35] De A F L, Pereira A D, Leal C D, et al. Effect of temperature on microbial diversity and nitrogen removal performance of an anammox reactor treating anaerobically pretreated municipal wastewater [J]. Bioresource Technology, 2018,258:208-219.
[36] Huang W, She Z, Gao M, et al. Effect of anaerobic/aerobic duration on nitrogen removal and microbial community in a simultaneous partial nitrification and denitrification system under low salinity [J]. Science of The Total Environment, 2019,651:859-870.
[37] Zhao J, Feng C, Tong S, et al. Denitrification behavior and microbial community spatial distribution inside woodchip-based solid-phase denitrification (W-SPD) bioreactor for nitrate-contaminated water treatment [J]. Bioresource Technology, 2018,249:869-879.
[38] Joo H S, Hirai M, Shoda M. Characteristics of ammonium removal by heterotrophic nitrification-aerobic denitrification byNo. 4 [J]. J. Bioscience and Bioengineering, 2005,100(2):184-191.
[39] Pai S L, Chong N M, Chen C H. Potential applications of aerobic denitrifying bacteria as bioagents in wastewater treatment [J]. Bioresource Technology, 1999,68(2):179-185.
[40] Shoda M, Ishikawa Y. Heterotrophic nitrification and aerobic denitrification of high-strength ammonium in anaerobically digested sludge bystrain No. 4 [J]. Journal of Bioscience and Bioengineering, 2014,117(6):737-741.
[41] Brazelton W J, Morrill P L, Szponar N, et al. Bacterial communities associated with subsurface geochemical processes in continental serpentinite springs [J]. Applied and Environmental Microbiology, 2013,79(13):3906-3916.
[42] Zhang P, Peng Y, Lu J, et al. Microbial communities and functional genes of nitrogen cycling in an electrolysis augmented constructed wetland treating wastewater treatment plant effluent [J]. Chemosphere, 2018,211:25-33.
[43] Ziegler M, Seneca F O, Yum L K, et al. Bacterial community dynamics are linked to patterns of coral heat tolerance [J]. Nature Communications, 2017,8:14213.
[44] Guimera R, Amaral L A N. Functional cartography of complex metabolic networks [J]. Nature, 2005,433(7028):895.
[45] Nesbø C L, Bradnan D M, Adebusuyi A, et al.. nov., sp. nov., the first described mesophilic species of the Thermotogales [J]. Extremophiles, 2012,16(3):387-393.
[46] Chen J, Han Y, Wang Y, et al. Start-up and microbial communities of a simultaneous nitrogen removal system for high salinity and high nitrogen organic wastewater via heterotrophic nitrification [J]. Bioresource Technology, 2016,216:196-202.
[47] Chen H, Wan J, Chen K, et al. Biogas production from hydrothermal liquefaction wastewater (HTLWW): Focusing on the microbial communities as revealed by high-throughput sequencing of full-length 16S rRNA genes [J]. Water Research, 2016,106:98-107.
[48] Mcilroy S J, StarnawskA A, Starnawski P, et al. Identification of active denitrifiers in full-scale nutrient removal wastewater treatment systems [J]. Environmental Microbiology, 2016,18(1):50-64.
Characteristics of bacterial community structure during the enrichment and domestication of heterotrophic nitrification-aerobic denitrification bacteria based on the typical city landscape water.
ZHOU Shi-lei1*, ZHANG Yi-ran1, SUN Yue1, YANG Wen-li1, HUANG Ting-lin2, LI Zai-xing1, LUO Xiao1, CUI Jian-sheng1, ZHOU Zi-zhen3, LI Yang3
(1.Pollution Prevention Biotechnology Laboratory of Hebei Province, School of Environmental Science and Engineering, Hebei University of Science and Technology, Shijiazhuang 050018, China;2.Key Laboratory of Northwest Water Resource, Environment and Ecology, Ministry of Education, School of Environmental and Municipal Engineering, Xi’an University of Architecture and Technology, Xi’an 710055, China;3.School of Energy and Environment, Zhongyuan University of Technology, Zhengzhou 450007, China) ., 2019,39(11):4831~4839
To explore the effects of different selective pressures on bacterial community structure during enrichment and domestication of heterotrophic nitrification-aerobic denitrification bacteria, bioinformatics analysis of samples taken from enrichment and domestication systems were carried out, using Miseq high-throughput sequencing. In detail,- and-diversity were examined, and network analysis was conducted. Proteobacteria, Bacteroidetes, Chloroflexi, Firmicutes, Actinobacteria, Cyanobacteria, and Acidobacteria were the main phyla identified. Meanwhile, the N-functional bacteria had an increased process. PCA (principal component analysis), NMDS (non-metric multidimensional scaling analysis) and PCoA (principal co-ordinates analysis) showed that microbial community structure was significantly altered with change in temperature, while the influence of different media was small. Network analysis indicated that module hubs and network hubs of bacterial communities were both rare taxa. VIF (variance inflation factor) and RDA (redundancy analysis) showed temperature, ammonia and nitrate were the most important factors affecting bacterial community function and composition. All results together indicate that Miseq high-throughput sequencing was an effective tool to explore changes in bacterial community structure during enrichment and domestication of heterotrophic nitrification-aerobic denitrification bacteria, which could in the future supply a reference to isolate “directional-accurate- efficient” microbial agents.
heterotrophic nitrification-aerobic denitrification;landscape water;Miseq sequencing;bioinformatics analysis;bacterial community structure
X172
A
1000-6923(2019)11-4831-09
周石磊(1987-),男,河北石家庄人,讲师,博士,主要从事好氧反硝化菌脱氮机理与调控机制的相关研究.发表论文27篇.
2019-04-28
国家自然科学基金资助项目(51909056);河北科技大学引进人才科研启动基金(1181278);河北省研究生创新资助项目(CXZZSS2018084)
*责任作者, 讲师, ZSLZhouShilei@126.com
