基于超高效液相色谱-高分辨质谱的非衍生化法测定面粉和燕麦中草甘膦及氨甲基膦酸残留
2019-11-07潘胜东童廷德叶美君陈晓红金米聪
潘胜东, 童廷德, 叶美君, 陈晓红, 金米聪
(1.宁波市疾病预防控制中心, 浙江省微量有毒化学物健康风险评估技术研究重点实验室, 浙江 宁波 315010; 2.赛默飞世尔科技(中国)有限公司, 上海 201206; 3.中华全国供销总社杭州茶叶研究院, 浙江 杭州 310016)
草甘膦(glyphosate, GLY)是一种高效广谱灭生性除草剂,广泛用于茶园、果园和麦田等,其生产量与市场销售额一直高居除草剂之首,约占全球除草剂市场份额的30%[1]。GLY及其代谢物氨甲基膦酸(aminomethylphosphonic acid, AMPA)能引起乙酰胆碱酶的活性降低[2]以及人体红细胞活性氧的增加[3]。此外,有研究表明GLY能引起实验大鼠的肝损伤[4]以及干扰牛的卵巢正常机能[5]。2017年GLY被国际癌症研究机构(International Agency for Research on Cancer, IARC)列入2A类致癌物[1]。近年来,美国孟山都开发了抗GLY转基因小麦并得到了美国监管部门的批准,该小麦植株可以耐受高浓度的GLY,为面粉中GLY残留埋下了安全隐患。2018年8月,美国环境工作组(EWG)对多个品牌的45份燕麦产品抽检,结果全部检出GLY残留,其中不乏许多知名品牌,一时间引起社会的广泛关注与忧虑。迄今,包括中国和欧盟(EU)在内的许多国家及国际组织对农产品中GLY的最大残留量(MRL)有着严格的限量规定,如GB 2763-2016《食品安全国家标准食品中农药最大残留限量》对小麦粉、全麦粉和小麦中GLY的MRL规定分别为0.5、5和5 mg/kg;欧盟(EU)对小麦、燕麦和荞麦中GLY的MRL规定分别为10、20和0.1 mg/kg[6],但我国未对燕麦中GLY的MRL加以规定。
目前,GLY及其代谢物AMPA的检测方法主要包括离子色谱(IC)法[7]、液相色谱-荧光(LC-FL)法[8]、气相色谱-质谱联用(GC-MS)法[9]和液相色谱-质谱联用(LC-MS)法[10-17]等。其中IC法抗干扰能力差,需要对样品提取液中大量杂质离子与目标分析物进行有效分离才能达到准确定量,具有成本高和实用性差等缺陷。LC-FL法测定GLY和AMPA需要配备柱后衍生装置,且用到巯基乙醇或巯基乙胺等气味较大的化学试剂,具有灵敏度差和假阳性率高等不足。GB/T 23750-2009采用GC-MS法测定植物性产品中GLY的残留量,该方法使用易挥发的衍生化试剂三氟乙酸酐,衍生化条件苛刻,不利于实验室的日常检测。随着LC-MS仪的普及,LC-MS法已被广泛应用于食品、环境样品和生物样品中GLY和AMPA的检测。由于GLY和AMPA的相对分子质量小、极性强,在常规的反相色谱柱上保留时间很短,因此目前大部分文献及SN/T 1923-2007《进出口食品中草甘膦残留量的检测方法液相色谱-质谱/质谱法》报道的LC-MS方法均采用9-芴甲基氯甲酸酯(FMOC-Cl)进行柱前衍生反应后再进行检测[11-13]。衍生化方法虽然能提高检测灵敏度和色谱保留,但衍生化操作过程较为繁琐,方法重现性差,在实际检测工作中应尽可能避免。SN/T1923-2007方法采用美国Bio-Rad CAX阳离子交换湿柱需要保证小柱不干涸,柱体积大、吸附容量小,且需采用常压过柱方式,洗脱液体积大,且含水量高,进一步浓缩耗时长,不利于实际工作的开展。虽有文献[13,14]报道基于LC-MS的非衍生化法直接测定GLY和AMPA,但多集中于基质简单的水样,局限性大,针对基质复杂的食品样品的报道相对较少。SN/T 4655-2016《出口食品中草甘膦及其代谢物残留量测定方法液相色谱-质谱/质谱法》采用LC-MS/MS非衍生化直接测定茶叶、玉米和稻谷等出口食品中GLY和AMPA,该法采用透析袋超声、RP(reversed-phase)柱和GCB(grafted carbon black)粉末进行3步净化过程,操作较为繁琐,且GCB属于非特异性吸附剂,对GLY和AMPA可能会有一定的吸附性,从而导致目标物的损失。江燕等[16]采用亲水作用色谱-串联质谱法直接测定稻米中的GLY和AMPA残留量,使用水提取后,用C18小柱和超滤膜净化,取流出液进行检测,该方法使用C18柱净化虽然能够除去疏水性干扰物,但难以除去提取液中离子型干扰物,不仅可能影响测定结果,还可能缩短氨基化色谱柱的寿命。周爽等[17]比较了MAX小柱、氨基柱和HLB小柱固相萃取小柱对植物性食品提取液净化性能的影响,结果表明,使用MAX小柱得到最优的结果,但该方法采用乙酸铵-乙腈的弱酸性流动相体系进行分离检测,峰形拖尾严重。
随着高分辨质谱技术的日趋成熟,精确分子量定性与定量所体现的优势为食品中有毒有害污染物的快速测定提供了有效的解决方案。本文采用混合阳离子交换固相萃取(MCX)净化,以氨基化高分子基HILIC柱为色谱分离柱,建立了基于超高效液相色谱-高分辨质谱(UPLC-HRMS)的非衍生化测定面粉与燕麦中GLY和AMPA残留的分析方法,可满足实验室日常监测的需求。
1 实验部分
1.1 仪器与试剂
Waters UPLC I Class型超高效液相色谱仪(美国Waters公司); Q-Exactive Orbitrap型高分辨质谱仪和Trace finder 3.3数据处理系统(美国Thermo Fisher公司); Legend RT型离心机(德国Heraeus公司); Sigma 1-14K高速冷冻离心机(德国Sigma公司);固相萃取装置(美国Agilent公司)。
LC-MS级乙腈和甲醇购自美国Thermo Fisher公司;HPLC级氨水购自德国Sigma公司;有证标准品草甘膦(GLY,纯度>98%)和氨甲基膦酸(AMPA,纯度>98%)购自德国Dr.Ehrenstorfer公司;有证内标物1,2-13C215N-GLY(100 mg/L)和13C15N-AMPA (100 mg/L)购自北京振翔科技有限公司。Oasis®MCX固相萃取小柱(150 mg/6 mL)、Oasis®PRiME HLB固相萃取小柱(150 mg/6 mL)、Waters ACQUITY UPLC HSS T3色谱柱(100 mm×2.1 mm, 1.8 μm)和Waters ACQUITY UPLC BEH C18色谱柱(100 mm×2.1 mm, 1.7 μm)购自美国Waters公司;ProElut GLY专用柱(6 mL)和Dikma Polyamino HILIC色谱柱(150 mm×2.0 mm, 5 μm)购自迪马科技。
1.2 实验方法
1.2.1样品前处理
将采集的燕麦采用搅拌机粉碎,过80目筛,面粉直接过80目筛,置于50 mL塑料离心管中,于-20 ℃保存,待测。
分别称取经粉碎与过筛的小麦粉和燕麦粉2.0 g(精确至0.01 g),置于50 mL离心管中,加入100 μL 10.0 mg/L 1,2-13C215N-GLY和13C15N-AMPA混合标准溶液,加入10 mL,水涡旋提取10 min,然后超声提取10 min,以8 500 r/min的速度离心5 min。取5 mL上清液,过经5 mL甲醇和5 mL水活化与平衡的MCX固相萃取小柱净化,弃去前3 mL流出液,收集后2 mL流出液。准确移取0.5 mL上述溶液于1.5 mL离心管中,加入0.5 mL乙腈,涡旋5 s,以16 000 r/min的速度离心5 min,过0.22 μm有机相滤膜,待测。
1.2.2色谱-质谱条件
色谱柱为Dikma Polyamino HILIC色谱柱(150 mm×2.0 mm, 5 μm);流动相A相为5 mmol/L pH 10.5乙酸铵溶液,B相为乙腈。梯度洗脱程序:0~2.00 min, 80%B; 2.00~3.00 min, 80%~10%B; 3.00~7.00 min, 10%B; 7.00~7.01 min, 10%~80%B; 7.01~10.00 min, 80%B;流速:0.3 mL/min;柱温:40 ℃;进样量:10 μL。
离子源:电喷雾离子源(ESI);扫描方式:负离子扫描;离子源温度:320 ℃;定量检测方式:平行反应监测模式(PRM);电喷雾电压:-3 000 V;脱溶剂化气压力:275.8 kPa;辅助气速率:180 L/h;射频棱镜电压(S-lens RF level): 60%;辅助气加热温度:300 ℃;分辨率:17 500;自动增益控制(AGC): 2×104;最大注入时间(Max IT); 150 ms;质荷比隔离窗口(isolation window): 1.0m/z。其他质谱参数列于表1。

表1 GLY及其代谢产物AMPA的质谱参数
* Quantitative ion pair.
1.2.3标准溶液的配制
1 g/L GLY和AMPA的单标准溶液:分别准确称取10.0 mg GLY和AMPA标准物质,用超纯水溶解、定容至10.0 mL。
10 mg/L GLY和AMPA混合标准溶液:分别准确吸取1.0 g/L GLY和AMPA的单标准溶液100 μL,用超纯水稀释至10 mL。
1.0 mg/L GLY和AMPA混合标准溶液:分别准确吸取10 mg/L GLY和AMPA的混合标准溶液1.0 mL,用超纯水稀释至10 mL。
10 mg/L 1,2-13C215N-GLY和13C15N-AMPA混合内标溶液:分别准确吸取0.5 mL 100 mg/L 1,2-13C215N-GLY和13C15N-AMPA内标溶液,用超纯水稀释、定容至5.0 mL。
系列标准溶液:准确吸取1.0 mg/L GLY和AMPA混合标准溶液50.0、100、250、500、1 000 μL于10.0 mL容量瓶中,加入5.0 μL 10 mg/L 1,2-13C215N-GLY和13C15N-AMPA,然后用超纯水稀释定容至刻度,配制成5.0、10.0、25.0、50.0、100.0 μg/L系列标准溶液,其中内标物质量浓度为25.0 μg/L。
1.2.4基质效应评价
分别准确称取6份2.00 g(精确至0.01 g)空白面粉和燕麦样品于50 mL离心管中,加入10 mL水涡旋提取10 min,然后超声提取10 min,以8 500 r/min的速度离心5 min。取5 mL上清液经MCX小柱净化,收集2 mL流出液,加入2 mL乙腈,涡旋5 s,以16 000 r/min的速度离心5 min,过0.22 μm有机相滤膜,收集样品溶液。采用净化后的样品溶液作为溶剂配制5.0、50.0和100.0 μg/L GLY和AMPA,同时加入两种内标溶液,使其终质量浓度为25.0 μg/L。取另5 mL未经净化的样品提取液,加入5 mL乙腈,涡旋5 s,以16 000 r/min的速度离心5 min,过0.22 μm有机相滤膜,收集样品溶液,采用未净化的样品溶液作为溶剂配制5.0、50.0和100.0 μg/L GLY和AMPA,同时加入两种内标溶液,使其终质量浓度为25.0 μg/L。采用50%(体积分数,下同)乙腈-水溶液作为溶剂,配制5.0、50.0和100.0 μg/L GLY和AMPA。通过UPLC-HRMS测定GLY和AMPA及其内标物的峰面积。分别作基质匹配和溶剂外标曲线和内标曲线。基质效应以η表示:η=(基质匹配标准曲线斜率-溶剂标准曲线斜率)/溶剂标准曲线斜率。其中,外标法基质效应ηex=(基质匹配外标曲线斜率-溶剂外标曲线斜率)/溶剂外标曲线斜率[18,19],内标法基质效应ηis=(基质匹配内标曲线斜率-溶剂内标曲线斜率)/溶剂内标曲线斜率。|η|值越大,说明基质效应越大,且η值正负分别代表基质增强和基质抑制作用。
1.2.5方法准确度与精密度实验
分别准确称取18份2.00 g(精确至0.01 g)空白面粉和燕麦粉样品于50 mL离心管中,加入100.0 μL 10.0 mg/L 1,2-13C215N-GLY和13C15N-AMPA内标混合溶液。分别准确加入10.0 mg/L GLY和AMPA的混合标准溶液各20、100和400 μL,配制成低、中、高3个水平(0.1、0.5和2.0 mg/kg)的加标样品,每个加标水平做6个平行试验。分别加入10 mL水,涡旋提取10 min,超声提取10 min,以8 500 r/min的速度离心5 min。取5 mL上清液经MCX小柱净化与乙腈沉淀蛋白质,采用UPLC-HRMS检测,每一浓度水平平行测定6次,计算回收率和精密度(RSD)。

图1 4种化合物质谱参数的优化结果
2 结果与讨论
2.1 仪器条件优化
2.1.1质谱条件优化
采用正负离子模式分别对GLY和AMPA及其同位素内标物进行一级扫描,发现目标物在ESI负离子模式下响应更高,准分子离子峰均为[M-H]-形式。因此,负离子模式下,确定了GLY、AMPA、1,2-13C215N-GLY和13C15N-AMPA的母离子分别为m/z168.006 7、110.001 2、171.010 5和112.001 6。经二级质谱图可知,4种化合物的二级质谱中均含有较高响应的碎片m/z62.964 2,选定其为定量离子。为提高检测灵敏度,分别对归一化碰撞能量(normalized collision energy, NCE)、Max IT、AGC和辅助气温度(auxiliary gas heated temperature, AUX gas temp)这4种质谱参数进行优化,结果见图1。
由图1a可知,NCE对质谱响应影响较大,当NCE较低时,GLY、AMPA、1,2-13C215N-GLY和13C15N-AMPA 4种化合物的质谱响应较低;随着NCE增大,质谱响应显著增大;当NCE达到一定值后,4种化合物的峰面积基本保持不变。经优化,分别选择GLY、AMPA、1,2-13C215N-GLY和13C15N-AMPA的NCE值为70、80、70和90。由图1b可知,Max IT对4种化合物的质谱强度有一定影响,变化趋势类似于NCE,先随Max IT值增大质谱响应增大,随后质谱响应趋于稳定。因此,4种化合物的Max IT值均取150 ms。由图1c可知,AGC对4种化合物的质谱响应影响不显著,后续实验设定AGC的值为2.0×104。AUX gas temp能显著影响化合物离子化效率,从而影响质谱响应。由图1d可知,随着AUX gas temp值的增大,4种化合物的峰面积逐渐增大;当AUX gas temp值为300 ℃时,质谱响应达到最大值,继续升高AUX gas temp值对质谱响应影响不大。考虑到节能和对仪器的损害最小化等因素,控制AUX gas temp值至300 ℃。
2.1.2液相色谱条件优化

图2 不同流动相组成对GLY色谱行为的影响
分别选择乙腈-水体系、乙腈-甲酸水溶液(pH=2.0)体系和乙腈-氨水溶液(pH=10.5)体系作为流动相,考察其对GLY和AMPA在Dikma Polyamino HILIC色谱柱上色谱行为的影响,结果见图2。当采用乙腈-水体系作为流动相时,GLY的峰形分叉,且灵敏度较低(见图2a);当采用乙腈-甲酸水溶液体系(pH=2.0)时,峰形较为对称,但色谱峰保留较差(GLY的保留时间tR=1.62 min),且质谱响应较差(见图2b);当采用乙腈-氨水溶液(pH=10.5)体系作为流动相时,GLY色谱峰形较为对称,色谱保留较好(tR=4.37 min),且质谱响应较好(见图2c)。AMPA在不同流动相体系中的色谱行为与GLY相似,因此后续的实验均采用乙腈-氨水溶液(pH=10.5)作为流动相体系。
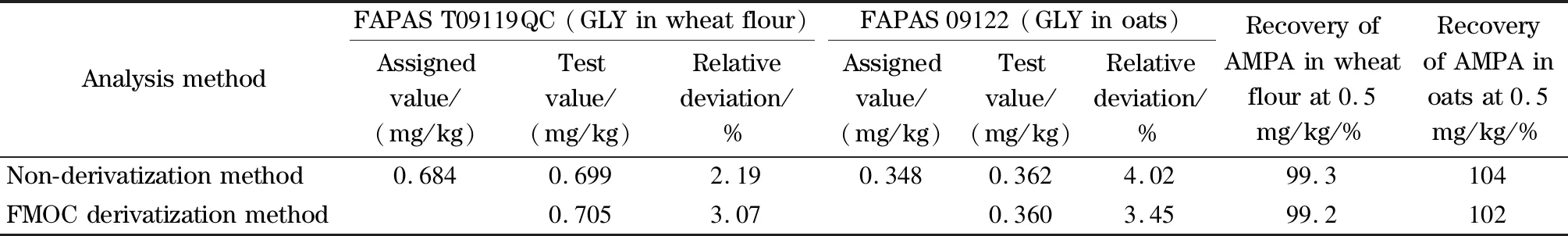
可能的原因是:(1)GLY的离子化方式为[M-H]-,碱性条件下能促进其离子化,从而提高质谱响应,反之,酸性与中性条件下不利于其离子化过程。(2)GLY分子存在3个pKa值,分别为分子中羧基(pKa1=2.34)、磷酸基团(pKa2=5.73)和氨基正离子(pKa3=10.9)的电离pKa值。当流动相pH=2.0时(乙腈-甲酸水溶液(pH=2.0)体系), GLY分子中羧基、磷酸基团和氨基正离子均未解离,此时体系中GLY分子存在形式单一,色谱峰形较为对称(见图2b)。当流动相2.0 图3 不同色谱柱对GLY的色谱行为的影响 如图3所示,考察了Waters BEH C18、Waters HSS T3和Dikma Polyamino HILIC 3种色谱柱对GLY和AMPA色谱行为的影响。当选择Waters BEH C18色谱柱时,GLY的峰形极差,拖尾现象严重(见图3a)。当选择Waters HSS T3色谱柱时,GLY峰形较窄,对称性好,但此时质谱响应较差(见图3b),这是因为Waters HSS T3色谱柱耐受pH范围为2.0~8.0,因此无法使用强碱性流动相,从而导致较低的质谱响应值。当选择Dikma Polyamino HILIC色谱柱时,GLY不仅色谱峰形对称、色谱保留佳,而且质谱响应也较高(见图3c)。因此,后续实验均采用Dikma Polyamino HILIC作为色谱分析柱。 图4 不同固相萃取小柱的净化效率对比 根据文献报道[11],采用纯水能较好地提取样品中的GLY和AMPA。本文不再对提取溶剂做过多的优化,实验过程均采用纯水作为提取溶剂。本文重点考察3种固相萃取小柱(PRiME HLB小柱、MCX小柱和GLY专用SPE柱)对GLY和AMPA的净化效果的影响,具体实验过程为:准确称取2.00 g面粉与燕麦粉样品于50 mL离心管中,加入10 mL去离子水,涡旋提取10 min,然后超声提取10 min,以8 500 r/min的速度离心5 min。取上清液,分别加入GLY、AMPA及其同位素内标物,使样品溶液中4种化合物的质量浓度为25.0 μg/L,然后分别采用PRiME HLB小柱、MCX小柱和GLY专用柱对其净化,通过比较各种固相萃取小柱对GLY与AMPA的回收率来评价净化效果,结果如图4所示。优化后3种固相萃取小柱的实验条件为:采用5 mL甲醇和5 mL水分别对3种固相萃取小柱进行活化与平衡,然后加入5 mL提取液过固相萃取小柱,舍弃前3 mL流出液,取后2 mL流出液。 由图4a可知,针对面粉与燕麦中的GLY,经水提取与乙腈沉淀蛋白质后(未经SPE柱优化)所得绝对回收率较低(<60%),而经稳定同位素内标校正后,面粉与燕麦中GLY的相对回收率有所提升(~80%),但还无法满足精确定量的要求,由此可见,提取液中存在干扰GLY及其同位素内标物的共提物,且对两者的质谱抑制强度不完全一致;经水提取与固相萃取小柱净化后,面粉中GLY的净化效果普遍比燕麦中好,且3种小柱对两种基质中GLY的净化效率排序为PRiME HLB小柱>MCX小柱>GLY专用柱。然而经同位素内标物校正后,PRiME HLB小柱的相对回收率明显提高,达到115%~120%; MCX小柱和GLY专用柱的相对回收率较为理想,分别为95.4%~102%和99.7%~109%,兼顾灵敏度和准确度的因素(MCX小柱绝对回收率明显高于GLY专用柱),最终选择MCX小柱作为测定面粉和燕麦中GLY的净化小柱。 由图4b可知,针对面粉与燕麦中的AMPA, PRiME HLB小柱和GLY专用柱的净化能力明显不足(绝对回收率<80%)。PRiME HLB小柱对两种基质中AMPA的绝对回收率与未经SPE净化的结果相当(均<80%),甚至GLY专用柱对AMPA的绝对回收率低于未经SPE净化的结果。相比之下,MCX小柱对面粉与燕麦中AMPA的净化能力最佳,绝对回收率分别为89.8%和83.8%,经同位素内标物校正后相对回收率分别为91.1%和82.8%。尽管经PRiME HLB和GLY专用柱净化的相对回收率尚且理想,但考虑到灵敏度以及与GLY同时检测的因素,最终选择MCX小柱作为测定面粉和燕麦中AMPA的净化小柱。 2.3.1线性方程和基质效应评价 由表2可知,针对面粉和燕麦样品中的GLY,未经SPE净化的基质效应相对较大,η值分别为-4.19%与-7.02%;经MCX小柱净化后,η值显著减小,降低至-0.23%与-2.67%。由此可见,MCX小柱净化能有效降低UPLC-HRMS检测GLY过程中的基质抑制效应。针对面粉和燕麦样品中的AMPA,经MCX小柱净化后,η值有一定程度减小,由未经SPE柱净化时的-5.27%与-6.18%降低至-2.29%与-5.38%。随后,本研究分别采用内标物1,2-13C215N-GLY和13C15N-AMPA进行校正,结果表明,面粉与燕麦中GLY和AMPA的|η|<3%,检测过程中基质效应可以忽略不计。因此,采用内标法和溶剂标准曲线法能准确定量检测面粉和燕麦中GLY和AMPA残留,且线性相关系数>0.999(见表2)。 表2 GLY和AMPA的线性范围、线性方程、相关系数、基质效应(η) 表3 GLY和AMPA在燕麦和面粉中的加标回收率和RSD(n=6) 2.3.2方法准确度和精密度 分别称取2.0 g面粉和燕麦粉空白样品,然后加入适量标准混合溶液,配制成低、中、高3个加标水平(0.1、0.5和2.0 mg/kg),按1.2.5节进行样品处理,采用UPLC-HRMS检测,每个加标水平测定6次,结果见表3。由表3可知,GLY和AMPA在低、中、高3个加标水平均有较好的准确度和精密度,GLY的加标回收率为93.8%~115%, RSDs为2.25%~9.54%; AMPA的加标回收率为89.8%~110%, RSD为2.45%~8.71%,能满足实验室日常监测的要求。 2.3.3方法检出限与定量限 本研究采用低浓度水平加标方式确定方法的检出限与定量限。分别称取2.0 g面粉和燕麦空白样品各3份,分别添加40、100和200 ng GLY和AMPA,使加标水平分别为0.02、0.05和0.1 mg/kg,按照1.2.1节过程处理样品,然后采用UPLC-HRMS进行分析,通过数据判断各低加标水平下是否会出峰,若有出峰,再通过仪器分析软件计算信噪比(S/N)。结果表明,当加标水平为0.02 mg/kg时(见图5a), GLY已出峰,S/N=25,AMPA未出峰;当加标水平为0.005 mg/kg时,发现在相同保留时间处GLY亦有出峰(S/N=5),因此确定GLY的检出限为0.005 mg/kg,定量限为0.02 mg/kg。当加标水平达到0.05 mg/kg时(见图5b), AMPA已出峰,软件计算S/N=4,确定AMPA的检出限为0.05 mg/kg,定量限0.1 mg/kg(见图5c)。与GB/T 23750-2009方法(GLY的LOQ为0.05 mg/kg)相比,本文对GLY的检测更灵敏。 图5 低水平加标样品的平行反应监测(PRM)色谱图 2.3.4进样系统残留控制 试验发现,当进一针高浓度GLY标准溶液或样品溶液后,进样系统会有一定程度的残留。为使得残留量降到最小,分别考察了纯乙腈、50%乙腈水溶液、纯水和0.5%甲酸水溶液作为强洗针液,控制较大体积洗针液体积(2 mL)。结果表明,0.5%甲酸水溶液作为洗针液时残留最少,但仍然无法消除残留现象,因此后续实验均采用0.5%甲酸水溶液作为洗针液。本研究选择中(50 μg/L)、高(100 μg/L)质量浓度GLY标准溶液进样(低质量浓度标准溶液进样未见明显残留),分别通过考察标准溶液与空白样品交替进样和进一针标准溶液后连续进空白样品两种方式计算GLY残留水平,以残留率(%)表示残留水平。残留率=纯水空白GLY峰面积/前一针标准溶液峰面积×100%,结果如图6所示,当进一针50 μg/L GLY标准溶液后,紧接着进一针纯水空白,然后继续进标准溶液和空白样品,如此交替12次试验,发现GLY的残留水平稳定在3.33%~3.92%之间,RSD为6.04%。当只进一针50 μg/L标准溶液后,连续进12针纯水空白样品时,随着空白进样次数增多,残留水平越来越小,且从进第2针空白溶液开始,残留水平降低至约1.0%,基本可以忽略,而进完第4针空白样品后进样系统中GLY已完全消除,进样100 μg/L标准溶液的残留与消除情况与此类似(见图6b)。此外,AMPA在进样过程中未发现明显的残留现象。因此,鉴于实际样品检测的实效性和经济性,在高浓度样品进样后,选择进一针空白纯水溶液,能有效减小进样系统GLY残留对定量检测结果的影响。 图6 进样系统GLY残留水平试验 2.3.5方法比对与实际样品分析 应用本研究建立的非衍生化测定方法与文献[10,11]报道的9-芴甲基氯甲酸酯(FMOC)衍生化方法分别对弗帕斯(FAPAS)面粉质控样(编号T09119QC,靶值为0.684 mg/kg,z值表示测定值与真值之间的偏离程度,|z|≤2.0的置信区间为0.452~0.916 mg/kg)和燕麦能力验证考核样(编号09 122,靶值为0.348 mg/kg)中GLY的测定结果进行比对。实验结果表明,两种方法对GLY的测定值十分接近(见表4),其中两种方法对面粉质控样的测定结果分别为0.699 mg/kg和0.705 mg/kg,与靶值的相对偏差分别为2.19%和3.07%;燕麦粉中GLY的方法比对结果表明,两种方法GLY的测定值分别为0.362 mg/kg和0.360 mg/kg,与靶值的相对偏差分别为4.02%和3.45%,能力验证结果表明,本方法对燕麦中GLY的测定结果满意(z=0.2, |z|≤2.0即判定合格)。由于FAPAS质控样品和能力验证样品中均不含AMPA,本研究采用空白面粉与燕麦样品加标回收方式(加标水平为0.5 mg/kg)对AMPA的两种测定方法进行比对与评价。结果表明,非衍生化法与FMOC衍生化法测定AMPA的回收率较为理想,分别为99.3%~104%和99.2%~102%。因此,本文建立的非衍生化法能准确测定面粉与燕麦样品中GLY和AMPA残留。应用本方法对市售的10份面粉与燕麦样品进行筛查与检测,结果未发现有GLY与AMPA。 表4 面粉与燕麦中GLY和AMPA的不同分析方法比对 FMOC: 9-fluorenylmethyl chloroformate; FAPAS: Food Analysis Performance Assessment Scheme. 本文建立了基于UPLC-HRMS的非衍生化法测定面粉与燕麦中草甘膦及其代谢物氨甲基膦酸的分析方法,该方法具有简便、快捷、灵敏和准确等优点,适用于实验室大批量样品的日常监测。

2.2 固相萃取柱的选择与评价
2.3 方法评价





3 结论