两种聚合酶在等位基因特异性引物延伸法检测SNP中的应用比较
2019-11-07陈载鑫谢岭平李柳燕许红丽方佩荣尹沃河付文金
陈载鑫 何 丹 谢岭平 李柳燕 许红丽 方佩荣 尹沃河付文金
1.广东医科大学附属东莞市厚街医院检验科,广东 东莞 523900;2.武岗市人民医院,湖南 武岗 422400
单核苷酸多态性(singlenucleotide polymorphism,SNP)是指在基因组水平上,特定核苷酸位置上存在2种或2种以上不同的碱基,且其中任何一种等位基因在群体中的频率大于1%[1]。SNP是一种单碱基水平的分子遗传标记,不仅可通过连锁或关联分析来定位疾病易感基因,而且有些SNP本身就可能会导致某些疾病或引起个体对药物反应的差异,同时,SNP图谱的建立还能有效地帮助破解人类生理学密码,了解人类进化的起源以及对患者进行有效的治疗。因此,SNP检测对疾病的风险性评估、诊断、预防和治疗等各方面均有很大的价值[2-4]。
目前有多种检测方法在SNP检测中成功应用,对完成相关基因检测或者药物靶向基因的研究提供了重要帮助,等位基因特异性引物延伸法(allele specific primer extension method,ASPE)结合测序技术是目前比较常见的检测方法之一,前者通过针对不同SNP位点基因型设计特异性引物达到特异性扩增和检测的目的[5-7],后者具有高通量、高速度、低成本、灵敏度高、重复性好、线性范围广、无需洗涤、组合灵活等特点,具有其他现有的检测技术无法取代的优势[8-9]。然而ASPE反应中聚合酶的选择会很大程度的影响结果的可靠性[10],因此研究不同聚合酶在等位基因特异性引物延伸法应用于SNP的研究具有重要的意义,为临床检测及应用提供数据参考。
1 材料与方法
1.1 一般材料
TaqTMHotStarDNA聚合酶和TspTMPlatinum DNA聚合酶及其相关试剂购自TAKARA公司,前者是一种创新型的抗体修饰的热启动酶。该酶在室温下活性被完全封闭,避免了在样品准备及第一循环反应升温阶段产生非特异性扩增和引物二聚体,增加DNA扩增的特异性。琼脂糖凝胶电泳法相关试剂及设备耗材购自BioRad公司。基因扩增仪购自中国透景生命科技有限公司;MagPlex-Tag微球和Luminex 200购自美国Luminex公司。
1.2 基因组提取
基因组提取采用人血液基因组提取试剂盒(Sangon,B518223),操作步骤按照试剂盒说明书进行:取300 μL抗凝全血加入600 μL Buffer TBP,充分混匀,室温放置1 min,然后8 000 r/min离心1 min,弃上清,用500 μL TE Buffer重悬沉淀,8 000 r/min离心1 min,小心弃掉上清,吸弃残液,加入180 μL Buffer Digestion和20 μL Proteinase K溶液,震荡混匀。56°C水浴30 min至混合液变澄清透明后加入 20 μL的 RNase A(10 mg/ml),加入60 μL Buffer PR,充分颠倒混匀,-20°C冰箱放置5 min,10 000 r/min离心5 min,将上清转移到新的1.5 mL离心管中,加入等体积的异丙醇,颠倒混匀,室温放置3 min后10 000 r/min离心5 min,弃上清,加入1 mL 75%乙醇,颠倒漂洗2~3次,10 000 r/min离心2 min,弃上清,开盖室温放置10 min,将乙醇残留挥发干净后加入50 μL TE Buffer溶解,提取的DNA取1 μL用于DNA浓度检测,其余样本置于-20°C保存。
1.3 引物设计
根据文献中报道的SNP的rs号在Genbank中查找CYP2C19*2突变点附近碱基序列,遵循普通PCR引物设计原则,用PrimerPlex软件设计高度特异性的ASPE引物,引物设计前都经过BLAST搜索排除同源区间,突变点位于设计引物的3′端,通过ASPE反应让扩增产物偶联捕获序列引物并延伸,本研究使用的引物序列如表1所示。
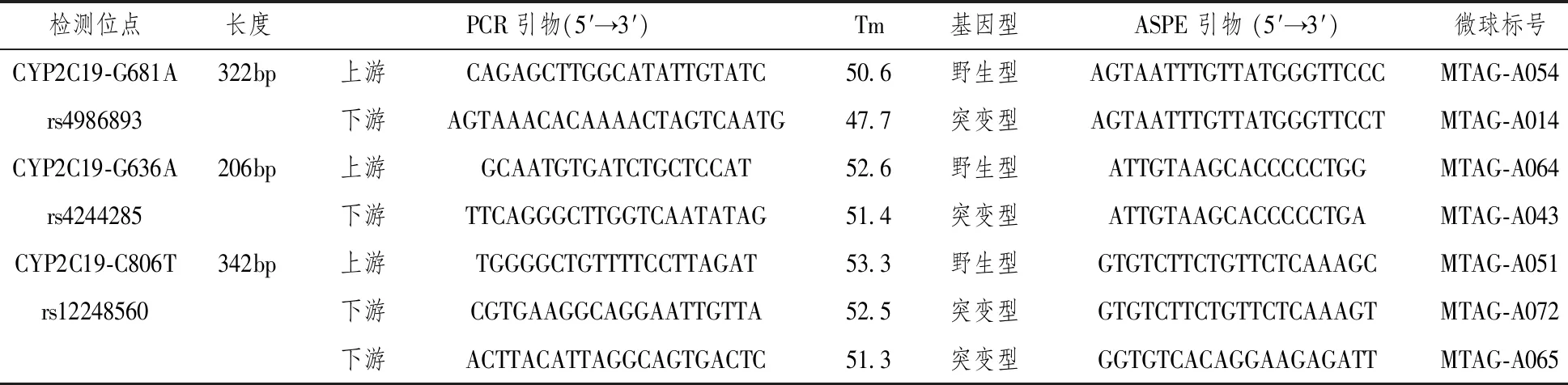
表1 本研究中用到的引物信息
1.4 多重PCR反应优化及产物纯化
为确保靶序列能同时有效扩增,首先我们对PCR反应体系的DNA模板浓度、引物浓度、退火温度及循环数进行调整。最终确定50 μL PCR体系:50 ng基因组DNA,10×PCR反应缓冲液(15 mM MgCl2, 500 mM KCl, 100 mM Tris-HCl, pH 8.3)5 μL,dNTP(各2.5mM)4 μL,上下游引物(10 μM)各0.5 μL,TaqTMHotStar DNA聚合酶1.5 U,无菌水30.5 μL。PCR循环参数:94℃预变性30 s;94℃ 30 s,57℃ 30 s,72℃ 30 s,5个循环;94℃ 30 s,55℃ 30 s,72℃ 30 s,30个循环;72℃延伸10 min。以无菌水作空白对照。留取部分PCR产物送广州艾基生物有限公司进行测序。PCR产物纯化:PCR产物5 μL加2.5 μL ExoSAP-IT混匀。37℃温育30 min以降解过量的引物和核苷酸(特别是dCTP),利于Biotin-dCTP在特异性引物延伸反应过程中有效延伸,随后在80℃温育20 min使酶失活。
1.5 ASPE反应及杂交反应的优化
前期研究中为确保ASPE引物进行有效延伸,对ASPE反应体系的引物浓度、MgCl2浓度和Biotin-dCTP进行调整;对退火温度进行52~58℃梯度设置确定最佳退火温度;杂交反应优化:配置1.5×TMAC杂交缓冲液(4.5 M TMAC,0.15%肌氨酸溶液,75 mM Tris-HCL, 6 mM EDTA,pH 8.0),稀释并混合16种MagPlex-TAG微球至每种微球终浓度为50个/μL,并涡旋或超声混匀微球约20 s。取ASPE反应产物2 μl混合至25 μL MagPlex-TAG微球混合液,96℃变性90 s,本实验将杂交温度分别设置37℃、45℃、50℃、55℃进行反应,在此温度下反应时间分别设定为:30 min、40 min、60 min。
1.6 数据分析和统计学分析
Luminex 200系统(安装xPonet软件)在37℃检测100 μL杂交混合液。检测系统输出中间荧光值(median fluorescence intensity, MFI)和等位基因MFI比率。等位基因MFI比率=MFI目标碱基/(MFI野生型+MFI突变型)。基因分型标准:等位基因MFI比率>0.75或<0.25为纯和野生型或纯和突变型,>0.25至<0.75之间为杂合突变型。采用Excel 2007软件进行变异系(CV)等统计学分析。
2 结 果
2.1 基因组质量检测
提取的基因组经过1%琼脂糖凝胶电泳检测,上样2.5 μL,结果如图1所示,说明本次提取的基因组没有出现降解或者RNA等污染的现象。取1 μL利用紫外分光光度计检测DNA浓度为420.8,886.8,534.6 ng/μL, OD260/280比值分别为1.80,1.82,1.85,说明本次提取的基因组均可以满足后续检测的使用要求,选择其中1号基因组样本用于后续试验。
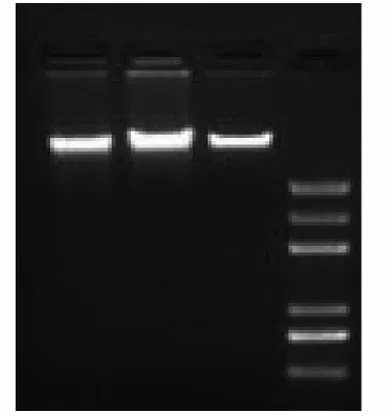
M:DNA marker;1,2,3:基因组
2.2 不同聚合酶敏感性验证
选择1号DNA样本进行后续研究,首先将1号样本稀释到200 ng/μL,然后进行倍比稀释,第一孔放置10 μL的1×TE缓冲液,取10 μL DNA样本加入后混匀,依次进行倍比稀释,最终样本浓度分别为12.5,6.25,3.13,1.56 ng/μL,每个样本取1 μL,然后经过1%琼脂糖凝胶电泳检测。结果如图2所示,在>6.15 ng/μL组中两种聚合酶均能检测到目的基因条带,在3.125 ng/μL样本组中,比较只有TaqTMHotStarDNA聚合酶有扩增条带,说明两种聚合酶的敏感性在较高浓度DNA样本下两者之间没有差异,但是在较低DNA浓度的情况下,TaqTMHotStarDNA聚合酶的扩增效果优于TspTMPlatinum DNA聚合酶。

1,2,3,4样本浓度依次为:12.5 、6.25、3.13、1.56 ng/μL
2.3 ASPE及杂交反应优化结果
本研究优化后的20 μL ASPE反应体系为:纯化后PCR产物2 μL,10× PCR反应缓冲液(15 mM MgCl2, 500 mM KCl, 100 mM Tris-HCl, pH 8.3)2 μL,Biotin-dCTP(400 μM)0.4 μL,dATP/dGTP/dTTP(各100 μM)1 μL,聚合酶0.6 U。最终优化ASPE反应条件:94℃ 预变性90 s;94℃ 30 s,57℃ 30 s,74℃ 1 min,10个循环;94℃ 30 s,55℃ 30 s,74℃ 1 min,30个循环;以无菌水作空白对照。
杂交反应优化结果:ASPE反应产物2 μL混合至25 μL MagPlex-TAG微球混合液,96℃变性90 s,本实验将杂交温度分别设置37℃、45℃、50℃、55℃进行反应,确定最佳杂交温度为37℃,在此温度下反应30 min、40 min、60 min,确定最佳杂交反应时间为40 min。
2.4 Luminex 200检测结果比较
本研究中 120份样本的基因测序结果显示其中有56例CYP2C19-681G/A,26例CYP2C19-681AA,16例CYP2C19-636G/A。TaqTMHotStarDNA聚合酶组检测结果与测序结果完全一致,MFI值范围从800到17 000,且背景值较低,范围从50到450,其中MFI/背景值比值均大于10,部分比值甚至超过90。TspTMPlatinum DNA聚合酶检测结果不能完全检测所有的基因型。
3 讨 论
SNP主要是指在基因组水平上由单个核苷酸的变异所引起的DNA序列多态性,是指在基因组上单个核苷酸的变异,包括转换、颠换、缺失和插入等形成的遗传标记,是人类可遗传变异中最常见的一种,大概占所有已知多态性的90%以上,因此国际人类基因研究学会花费大量的资金来完成人类单体型图谱。SNP分析从根本上来说其实就是确定一对染色体的每种基因的两个拷贝,哪种点突变在这两个拷贝中存在,因此利用定量PCR方法,并配合其他检测技术可以很好的检测SNP结果。常规SNP检测如DNA测序、限制性酶切片段长度多态性(RFLP)、单链构象多态性(SSCP)、ASPE等均可以有效检测SNP。ASPE中引物延伸是3’端开始的,所以3’末端的碱基对引物的延伸至关重要。如果这个碱基与模板互补,则引物可以进行不间断延伸,PCR可以正常进行,得到特定长度扩增带。反之,则不能延伸。所以只要将突变与正常等位基因所不同的碱基安排在3’最末端,当用某一含突变序列的引物进行PCR时,如果得到特异扩增带,表明被测基因含有该种突变,反之则表示没有突变。
DNA聚合酶在基因扩增中具有重要的作用,聚合酶的选择会直接影响检测的准确性,目前研究中低保真酶和高保真酶使用仍比较广泛,因此本研究选择检测中常用的两种酶验证其在等位基因特异性引物延伸法检测SNP中的效果,结果显示高保真酶的敏感性与低保真酶相比较在高浓度DNA样本下两者之间没有差异,但是在较低DNA浓度的情况下,高保真酶的扩增效果优于低保真酶组。在一般PCR实验中,低保真酶组和高保真酶组均能扩增出长度一致的目的基因条带,但是出现错配和基因碱基缺失的概率明显高于高保真酶组。这可能是由于高保真酶具有3′→5′外切酶活性有关,本研究与陈琳玲等的研究结果一致[10-11]。同时有研究指出在短片段扩增过程中目的基因的长度也会影响扩增效果,在小于1 000 bp的目的基因扩增中两种酶之间没有差异[12-13]。
本研究结果显示,利用ASPE法结合Luminex 200平台进行SNP检测时,聚合酶的选择也会在一定程度上影响检测结果,我们实验结果表明在TaqTMHotStarDNA聚合酶和TspTMPlatinum DNA聚合酶之间进行比较,前者在应用中的分辨率明显高于后者,因此,在临床中为了检测的可靠性我们推荐使用TaqTMHotStarDNA聚合酶。
本研究还存在一些不足,如本研究只是针对特定的基因型进行检测,人类基因组的结构是比较复杂的,该方法能否对所有基因进行无差错的检测还不得知,另外本实验中只对这两种聚合酶的效果进行了比较,尚不确定其它聚合酶是否会有更加好的效果,还需要进一步实验验证。
