Fe1(OH)x-Pt界面单位点催化剂在PROX反应中展现出高催化性能
2019-11-04包信和
包信和
1中国科学院大连化学物理研究所,催化基础国家重点实验室,2011-能源材料化学协同创新中心,辽宁 大连 116023
2中国科学技术大学,合肥 230026
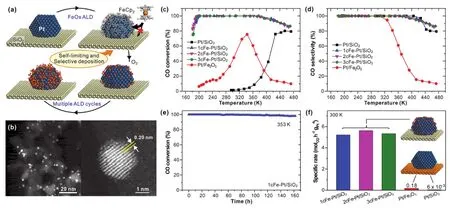
(a)基于原子层沉积技术的Fe1(OH)x-Pt界面单位点的精准构筑与调控示意图。(b) Fe-Pt/SiO2样品的STEM电镜图。xcFe-Pt/SiO2, Pt/SiO2和Pt/Fe2O3三种催化剂在PROX反应中的CO转化率(c)和选择性(d)对比。(e)在CO2和水汽同时存在情况下,1cFe-Pt/SiO2单位点界面催化剂在PROX反应中的稳定性,反应温度为353 K。(f)上述催化剂在300 K反应温度下的比质量活性对比。
负载型金属纳米催化剂在工业催化中有着广泛的应用,在该类催化体系中,金属-氧化物(或氢氧化物)界面扮演着重要的角色1–6。因此,实现金属-氧化物(或氢氧化物)界面的最佳构建一直是提高负载型金属催化剂催化活性的有效途径。目前,该类界面结构调控一般采用两种途径:一是减少金属纳米颗粒尺寸,使其达到至亚纳米,甚至单原子范围,增加金属与氧化物载体的接触7,8。在该方法中,伴随的量子尺寸效应往往也会对界面性能产生显著影响9,此外,金属颗粒以及单原子的稳定性和负载量等也是影响催化剂综合性能的重要因素10;二是利用氧化物(或氢氧化物)包裹金属纳米颗粒,使界面由颗粒与载体的二维局部接触转化成三维全方位接触,一方面最大程度地增加界面,同时实现界面位点的优化和性能增强5,11,12。因此,精准调控氧化物(氢氧化物)在金属颗粒表面的覆盖度和包裹形态成为了实现界面特性调控的关键。然而,在原子级精准条件下实现金属-氧化物(或氢氧化物)界面的可控构建以及反应气氛下的界面结构表征目前仍然存在巨大挑战。
最近,中国科学技术大学路军岭课题组利用二茂铁分子在Pt金属表面自限制式的解离吸附以及利用表面吸附物种空间位阻效应,创造性地运用原子层沉积(ALD)技术,成功地在SiO2负载的Pt金属纳米颗粒表面上精准构筑出原子级Fe1(OH)x物种,进而促成了新型Fe1(OH)x-Pt界面单位点催化活性中心的形成。在富氢条件下的CO优先氧化(PROX)反应中,该催化剂表现出很高的催化活性和优良的稳定性,在-75 °C至107 °C的温度区间内,成功实现了100%选择性地去除CO。实验结果显示,该催化剂表现出的比质量催化活性(5.21 mol·h-1·g-1)是Pt1/FeOx单原子催化剂的8倍,是传统Pt/Fe2O3催化剂的30倍。中国科学技术大学韦世强教授课题组利用原位X射线吸收谱(XAFS)技术,从实验上准确证实在PROX反应气氛中涉铁物种的配位结构为Fe1(OH)3,并进一步发现该物种具有高还原特性,在室温就能被氢气还原为Fe1(OH)2,很好地揭示了其低温条件下高催化活性的内在原因。中国科学技术大学杨金龙教授课题组采用密度泛函理论(DFT)方法确定了Fe1(OH)3在Pt表面上的空间构型,从理论上证实Pt颗粒表面上形成的Fe1(OH)3-Pt单位点界面是其催化活性中心,进一步揭示了该催化反应过程的分子机理:吸附的CO首先进攻Fe1(OH)3-Pt体系中的一个OH,形成COOH表面中间物种,随后,O2在该界面处以极低的势垒发生活化,形成的原子O进攻COOH,生成CO2从催化剂表面脱附。
该工作近期在Nature杂志上在线发表13,报到的这种具有高活性和高稳定性的Fe1(OH)x-Pt界面单位点新型催化剂结构和相关的反应机理为人们设计高活性金属催化剂提供了一个新思路。
