合成β-内酰胺酶抑制剂阿维巴坦的关键中间体的研究进展
2019-11-01陆凡武红丽沙凤曹祁陈姣曹飞韦萍
陆凡 武红丽 沙凤 曹祁 陈姣 曹飞 韦萍
(南京工业大学生物与制药工程学院,南京 211816)
自抗生素被发现并应用于医学领域后,许多感染性疾病得到了有效控制,新生儿死亡率及手术后感染率大大降低,因此延长了人类生存的平均寿命。然而,细菌的耐药性也随之产生,人类的健康和生命安全又面临新的考验[1]。当前,面对日趋严峻的“抗菌”形势,一些国家已经开始采取激励措施,鼓励新抗生素的研发。美国于2012年7月通过了《鼓励开发抗生素法案》(GAIN),根据规定,符合标准的抗生素药物将获得额外5年的市场独占权,以帮助开发者收回投资。然而新抗生素的开发难度越来越大,因此通过复方抗生素来提升治疗效果也是解决细菌耐药性的途径之一[2]。
β-内酰胺酶抑制剂阿维巴坦(2)本身几乎不具有抗菌活性[3],但其抑酶谱广,与其他抗生素联用可有效恢复或增强其活性。2015年2月,由阿特维斯与阿斯利康联合研发的阿维巴坦与头孢他啶组成的复方药物avycaz获得美国食品药品监督管理局(FDA)批准上市,用于治疗成人复杂性腹腔内感染及复杂性尿路感染等系列疾病[4-5]。阿维巴坦属于DBOs类化合物,化学名为[(1R,2S,5R)-2-(氨基羰基)-7-氧代-1,6-二氮杂双环[3.2.1]辛-6-基]硫酸单酯。其合成路线非常繁多,起始原料大致可分为L-焦谷氨酸衍生物、手性的哌啶环衍生物等,而以L-焦谷氨酸衍生物为起始原料的合成路线大都需要经历开环后增碳再闭环形成哌啶环衍生物的步骤[6]。因此,人们开始研究直接以手性的哌啶环衍生物为原料的合成路线。2014年,默克公司的一个研发小组报道了以手性哌啶环衍生物顺式-5-羟基哌啶-2-甲酸(cis-5-hydroxy-L-pipecolic acids,cis-5-hypip,1)为原料,合成同样具有二氮杂二环辛烷基本骨架的化合物,总收率高达39%,且该工艺路线具有公斤级放大规模的潜力[7]。而从化学结构上来看,阿维巴坦具有两个手性中心,这两个手性中心都在哌啶环上,因此以cis-5-hypip作起始原料,则可较好地解决手性光学纯度问题(图1)。cis-5-hypip同样也是TNF-α转化酶抑制剂[8]合成的重要中间体。其本身也对导致存储谷物腐败的曲霉菌具有抑制作用[9]。目前,对cis-5-hypip合成的研究,主要分为化学合成和酶促合成两个方面。

图1 cis-5-hypip(1)与阿维巴坦(2)Fig.1 cis-5-hypip(1)and avibactam(2)
1 化学合成cis-5-hypip
Bailey等[10-11]以谷氨酰胺(3)为原料,将其转化为N-苄氧基羰基甲酯(4)后,用亚硝酸叔丁酯作为[NO]+的来源并在乙腈中进行处理,使其侧链酰胺基团选择性水解的同时没有水解酯或氨基甲酸酯,从而得到光学纯的N-苄氧基羰基-L-谷氨酸-α-甲基酯(5)(Z-Glu-OMe,Z=苄氧基羰基)(74%)。用氯甲酸乙酯处理(5)后与混合酸酐和重氮甲烷反应生成重氮甲酮(6)(收率82%),接着通过碳烯引入N-H键将(6)转化为受保护的5-氧代哌啶酸(7)(75%)。最后用硼氢化钠立体特异性还原得到cis-5-hypip衍生物(8)(93%),脱保护,得到最终产物(1)(图2),总收率为42%。但是该方法有使用工业上难以使用的重氮甲烷的工序,因此难以工业化,还存在所得到的化合物为立体异构体的混合物的问题。
Krishnamurthy等[12]以具有叔丁氧基羰基(Boc)保护的丙二酸二乙酯(9)为原料,在乙醇钠中用过量的4-溴-1-丁烯处理(9),得到烷基化二酯,接着用固体LiOH选择性皂化,得到半酸半酯,在回流甲苯中脱羧,得到(RS)-10(相对于9的总收率为59%)。用α-胰凝乳蛋白酶处理后得到纯的对映体2-氨基-5-己烯酸衍生物(R)-10(51%)、(S)-11(46%)。在Et3N中用乙基溴将(S)-11酯化,得到(S)-10,产率84%。接着用过量的m-CPBA进行一系列处理得到环氧化物(12)(产率为90%)。再将非对映异构环氧化物(12)分离得到手性二醇(2S,5S)-13和手性环氧化物(2S,5R)-12,收率分别为46%和47%。其中手性环氧化物(2S,5R)-12经过6步反应最终可以得到反式-5-羟基哌啶-2-甲酸。而手性二醇(2S,5S)-13在催化剂的作用下用TsCl处理二醇的选择性甲苯磺酰化得到单甲苯磺酰化产物(14)。使用NaI将(14)中的甲苯磺酰基基团用碘基团取代,得到(2S,5S)-15(95%)。接着除去Boc基团使分子内环化后用Boc基团再次重新保护氨基酸的氮,得到(2S,5S)-16(76%),最后将其转化为(1)(图3)。该方法需要两次分离非对映异构体,每次分离都只能有一半的收率,导致最终总收率更低(7%),且分离步骤较复杂,很难实现工业化生产。

图2 cis-5-hypip的化学合成路线一[11]Fig.2 Chemical synthetic route of cis-5-hypip[11]
2016年,竹原润等[13]以(3S)-4-氯-3-羟基丁酸乙酯(17)为原料,先将羟基进行保护得到(18),接着将(18)中的酯基还原从而合成(3S)-4-氯-3-(四氢吡喃-2-基氧基)-丁烷-1-醇(P=四氢吡喃基)(19),对它的羟基进行磺酸酯化从而得到(3S)-甲磺酸-4-氯-3-(四氢吡喃-2-基氧基)-丁酯(20),将其与乙酰基氨基丙二酸二乙酯(21)反应后得到(22),环化,脱保护,得到(5S)-1-乙酰基-5-羟基-哌啶-2,2-二甲酸二乙酯(24)。接下来将(24)中的酯基水解,通过使一个羧基与羟基反应而进行内酯化,然后使羧基脱羧得到(26);或者将酯基水解,通过将一个羧基脱羧而得到2位单羧酸的立体异构体混合物(25),接着使该立体异构体混合物进行异构化内酯化得到(26)。最后将(26)中的内酯水解,并使其中的酰胺键断裂,从而得到顺式-5-羟基哌啶-2-甲酸,总产率达到44.8%(图4)。该方法避免了因立体异构体拆分导致产物较大损失的问题,具备工业化生产的潜力。
2 酶促合成cis-5-hypip
由于化学合成需要分子内环化生成哌啶环且需要复杂的手性拆分方法,因而导致较长的合成步骤,使得总产率较低。而酶具有高度选择性和专一性的特征,因此,人们也开始研究通过生物法制备cis-5-hypip。不同于化学合成中需要进行分子内环化形成哌啶环,生物法可以运用酶的立体和区域选择性,直接在L-哌啶甲酸(L-pipecolic acid,L-Pip)的5位碳上进行羟基化,这其中的关键酶为哌啶甲酸羟化酶(pipecolic acid hydroxylases,PiHs)。2016年,Hibi等[14]发现了来自于Fusarium oxysporumc8D和Aspergillus nidulansFGSC中的两种顺式-4-哌啶甲酸羟化酶基因(FoPip4H和AnPip4H),可以将L-Pip羟基化生成顺式-4-羟基哌啶甲酸。2017年,Huttel等[15]从Streptomycessp.和Frankia alni中发现了两种PiHs(GetF和PiFa),但是这两种酶只能对L-Pip的3位碳进行羟基化,5位羟基化的哌啶甲酸羟化酶至今还没有被发现。而在之前的报道中,脯氨酸羟化酶(proline hydroxylases,PHs)具有较为宽广的底物选择性,可以催化L-Pip生成羟基哌啶甲酸[16-17],如顺式-4-脯氨酸羟化酶(cis-proline-4-hydroxylases,cis-P4H)可以将L-Pip羟基化生成cis-5-hypip。为了降低成本,人们又以L-赖氨酸(L-lysine,L-Lys)为原料,通过赖氨酸环化脱氨酶(lysine cyclodeaminase,LCD)生产L-Pip,最终形成了从L-Lys到L-Pip再到cis-5-hypip的合成过程(图5)。

图3 cis-5-hypip的化学合成路线二[12]Fig.3 Chemical synthetic route of cis-5-hypip[12]
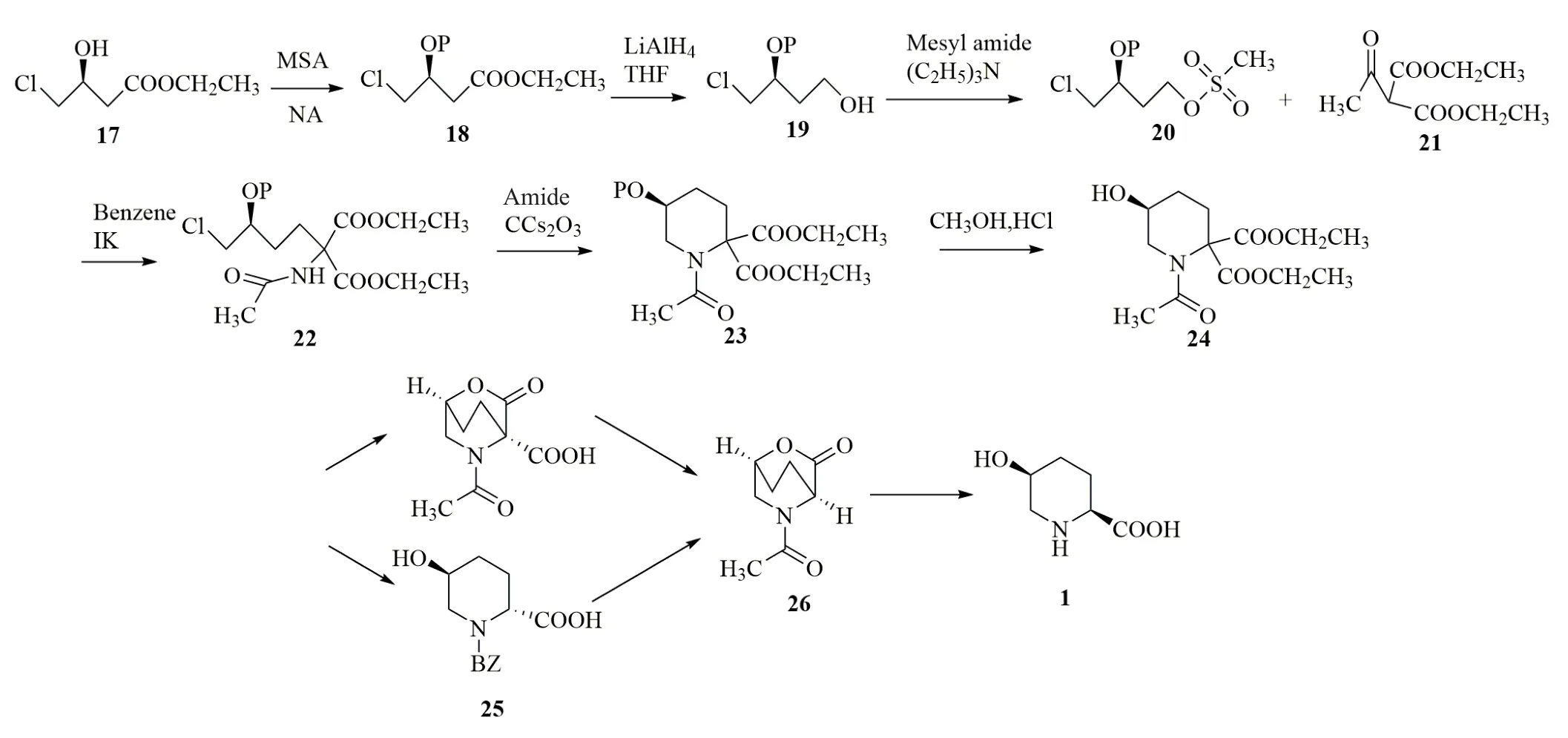
图4 cis-5-hypip的化学合成路线三[13]Fig.4 Chemical synthetic route of cis-5-hypip[13]
2.1 赖氨酸环化脱氨酶
L-哌啶甲酸也是合成许多重要药物的手性中间体,如卡因类局麻药物S-利多卡因、S-罗哌卡因、S-布比卡因、S-左布比卡因等[18]。目前,以L-Lys脱去α位氨基来实现脱氨环化反应过程有3条,分别是①L-赖氨酸-α-氧化酶与Δ1-哌啶-2-甲酸酯(Δ1-piperideine-2-carboxylate,P2C)还原酶途径;②L-赖氨酸-2-氨基转移酶与P2C还原酶途径;③L-赖氨酸环化脱氨酶途径[19]。由于前两种途径都是先生成一个P2C中间产物后再由P2C还原酶催化生成L-PA,反应需要两种酶参与且需要分两步完成,因此人们更加倾向于更高效,更简便的赖氨酸脱氨环化酶途径,其在NAD+的帮助下,可以直接将线性底物L-赖氨酸催化环化成为L-哌啶甲酸,相较其他途径,更为简便快捷[20]。

图5 cis-5-hypip的生物合成过程Fig.5 Biosynthesis route of cis-5-hypip
赖氨酸环化脱氨酶(LCD)的发现要归功于对著名的移植抗排异药物雷帕霉素的研究[21]。Moluar等[22]在研究链霉菌中次级代谢产物雷帕霉素的合成过程时,通过同位素标记意外地发现雷帕霉素中的效价基团哌啶甲酰胺来源于初级代谢产物赖氨酸,他们通过基因挖掘技术,初步获得了LCD的基础信息。然而早期对于LCD的研究因为技术手段的匮乏而十分局限,甚至出现了误解(认为LCD也可以催化D-赖氨酸产生D-哌啶甲酸)。2006年,Gatto等[23]对来源于Streptomyces hygroscopicus的赖氨酸环化脱氨酶的基因rapL的酶学性质进行了全面的表征并成功的将rapL在大肠埃希菌中进行了异源表达,同时也对ShLCD的辅酶依赖性做了详细的考察,在外源性NAD+存在下,初始速率提高8倍,他们将NAD+进行催化利用的过程被称之为“复杂NAD+依赖型转化”(complex NAD+-dependent transformation),丰富了改造这类酶的经验和基础。2018年,Ying等[24]对以来源于Streptomyces pristinaespiralis的赖氨酸环化脱氨酶SpLCD进行蛋白质结晶条件的筛选与优化,通过X射线衍射法解析SpLCD及其与小分子配体复合物的三维晶体结构。进而对SpLCD进行分子动力学模拟,并结合定点突变阐述SpLCD与底物的识别与转运机制、产物的释放机制,筛选出了组合突变株Val61-Val94-SpLCD,相比野生型SpLCD,其催化效率提高了1.81倍,并且解除了底物与产物抑制。随后利用该组合突变株进行了全细胞催化合成L-Pip的研究,在单次底物上载量为50g/L,分批补料3次的条件下,使最终的L-Pip产物浓度达到了73.4g/L,展现了一定的实际应用潜力。
2.2 脯氨酸羟化酶
由于目前尚未发现对L-Pip上5位碳进行羟基化的PiHs,而PHs具有羟基化L-Pip的能力,因此人们转向对PHs的利用与改造[17]。PHs属于非血红霉素Fe(Ⅱ)/α-酮戊二酸(α-KG)双加氧酶家族,这些酶通过与Fe2+、α-KG和O2产生的高反应性铁基(FeIV=O)复合物将羟基引入底物上没有反应活性的碳,是具有高度的区域和立体选择性的C-H催化反应[25],与NAD(P)H依赖性P450单加氧酶相反,它们的共底物α-KG价格便宜,因此不需要再生循环系统。根据不同的区域和立体选择性,PHs可以分为4种类型:顺式-3-、反式-3-、顺式-4-和反式-4-脯氨酸羟化酶(cis-P3H、trans-P3H、cis-P4H和trans-P4H),分别将L-Pip羟基化形成对应的羟基哌啶甲酸(图6)。
1996年,Lawrence等[26]成功从绿灰菌素生产者Streptomyces griseoviridusP8648中发现了第一个PHs:trans-P4H。Sankyo公司的科学家使用Heliocerus oryzae和Acrocylindrium oryzae生产顺式-4-羟基脯氨酸,最大产量为30mg/L[27]。Mori等[28-29]使用高灵敏度的HPLC分析筛选了超过3000种放线菌菌株的脯氨酸羟化酶活性,找到了8株具有trans-P4H活性的菌株,还鉴定了4种具有cis-P3H活性的菌株,之后,Mori等[29-34]还对这些基因进行PCR扩增和克隆并构建重组表达载体,以大肠埃希菌为宿主进行异源表达,为之后进一步对这类酶的研究打下了基础。2009年,Hara等[35]报道了从Sinorhizobium meliloti和Mesorhizobium loti的基因组中鉴定出的cis-P4H,并将这些cis-P4H分别命名为SmP4H和M1P4H,但是这些酶在催化L-Pip生成羟基哌啶甲酸的过程中会产生出两个区域异构体cis-5-hypip和cis-3-hypip,为了选择性地生产cis-5-hypip,他们对SmP4H进行了3轮定向进化并成功地创建了cis-P4H三重突变体V97F/V95W/E114G,大大增加了对cis-5-hypip的区域选择性(图7),同时还提高了酶的催化活性,使cis-5-hypip的产量达到778mg/L[36]。Haibin等[37]同样使用蛋白质工程对SmP4H进行改造,但即使改变了SmP4H的基因,也会生产出约9%的cis-3-hypip。Keisuke等[38]报道了来自于Swgniliparus rugosus的cis-P4H(SrP4H),其在将L-Pip羟基化生成cis-5-hypip的过程中,只生成2%的cis-3-hypip,但是这种酶的催化活性较低,产量只有50mg/L。Ryoma等[39]从Xwnorhabdus doucetiaeFRM16和Xwnorhabdus romaniistr.PR06-A中鉴定出两种具有很高5位羟基化选择性的PHs(XdP4H和XrP4H),其中XdP4H只生产出0.17%的cis-3-hypip,同样XrP4H也只生产1.75%的cis-3-hypip,但是这两种酶同样存在催化活性低的问题。洪浩等[40]筛选了现有的PHs序列的同源序列,筛选出一个来自于Kordia jejudonensis的序列,具有较强的催化活性,他们对该羟化酶的氨基酸序列进行了突变试验,得到了多株具有更高5位选择性的突变株,在催化过程中3位的异构体只占到2%左右。
3 小结
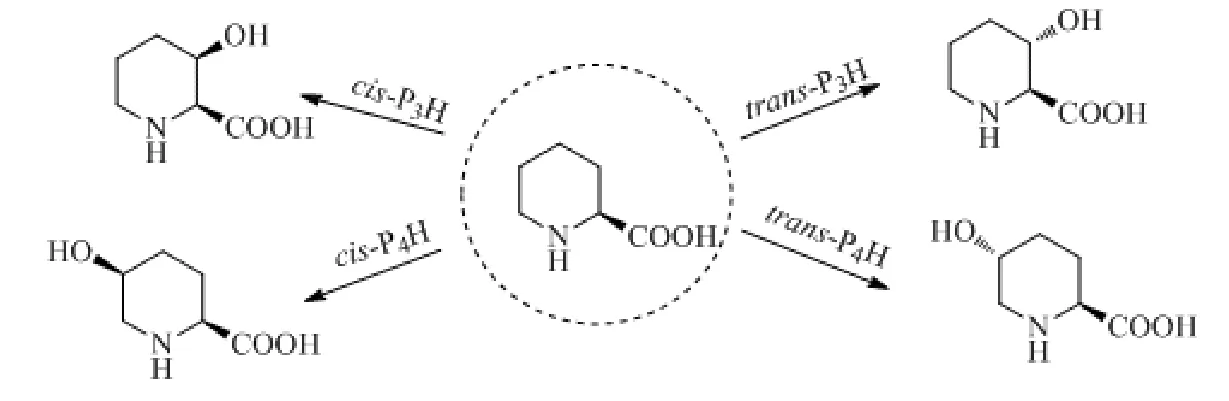
图6 PHs的类型[17]Fig.6 The different kind of PHs[17]

图7 PHs的定向进化[36]Fig.7 Directed evolution of PHs[36]
随着细菌耐药性的问题越来越严重,阿维巴坦作为一个能与其他抗生素联用并提升疗效的广谱抑酶制剂,必将会受到广泛应用。而对于其合成路线中的关键中间体cis-5-hypip的研究也将越受关注。相对于化学合成复杂的手性拆分,导致产率较低,生物方法更加直接与高效,但是目前生物催化也存在一些问题,如SpLCD在催化底物转化过程中酶活力不断下降,并逐渐开始产生副产物P2C,而PHs在催化过程中的催化活性和选择性也不是很好,这些都是在工业化生产之前需要解决的问题。无论是酶促合成或化学合成的方法,都期待更多、更高效、更利于工业化生产的cis-5-hypip合成路线的问世。
