Cellular models for human cardiomyopathy:What is the best option?
2019-10-31NereaJimenezTellezStevenGreenway
Nerea Jimenez-Tellez,Steven C Greenway
Nerea Jimenez-Tellez,Department of Biochemistry & Molecular Biology,Cumming School of Medicine,University of Calgary,Calgary,AB T2N 4N1,Canada
Steven C Greenway,Departments of Pediatrics,Cardiac Sciences,Biochemistry & Molecular Biology,Cumming School of Medicine,Libin Cardiovascular Institute of Alberta,Alberta Children’s Hospital Research Institute,University of Calgary,Calgary,AB T2N 4N1,Canada
Abstract
Key words: Cardiomyopathy; Mitochondria; Induced pluripotent stem cells; Fibroblasts;Cellular models
INTRODUCTION
The cardiomyopathies are defined as a group of diseases of the heart characterized by abnormal structure and function of the myocardium[1].The cardiomyopathies have been classically grouped according to cardiac morphology with the major categories being:hypertrophic cardiomyopathy (HCM),restrictive cardiomyopathy,dilated cardiomyopathy (DCM),arrhythmogenic right ventricular cardiomyopathy and left ventricular non-compaction cardiomyopathy (LVNC)[2].These groups can be further subdivided into genetic and acquired forms based on disease mechanism[2].The genetic cardiomyopathies generally arise in childhood or early adulthood and include metabolic and monogenic diseases.
The inborn errors of metabolism (IEM) are a heterogeneous group of rare genetic diseases caused by defects in energy production or intermediary metabolism[3,4].Within the pediatric cardiomyopathies,IEM affect between 5% and 26% of infants and children[5].There are more than 40 different IEM that are associated with the development of cardiomyopathy[3].The mitochondrial cardiomyopathies represent the largest subset and result from pathologic mutations in either mitochondrial or nuclear genes[6]that ultimately lead to dysfunction of the electron transport chain[7],the main supplier of cellular energy under aerobic conditions[8].Since the heart is one of the most energy-demanding organ in the body[9],cardiomyopathies are found in 20%-40% of children with mitochondrial disease[10].Given the early onset of these devastating multisystem diseases,research into disease mechanism and the identification of potential therapeutics is essential.However,the heterogeneity and rarity of the IEM and the mitochondrial cardiomyopathies preclude randomized clinical drug trials with standardized end-points.This makes disease modelling using animals or cells an essential component in the study of these diseases.
ANIMAL MODELS
The use of animal models for research,with rodents in particular,continues to represent the most commonly used and successful approach in reductionist biology.However,despite its many successes,this methodology is still questioned because of ethical implications,the frequent inability to totally recapitulate human genetic variability[11]and the fact that important species-specific differences exist for many aspects of biology which complicate both the study of disease and the translation of therapies into human subjects[12].For example,in cardiac research specifically,the use of rodent models may be limited due to substantial biological differences in the cardiovascular system between rodents and humans.Rodent hearts beat at considerably higher heart rates (200-300 beats per minute) than humans (60-100 beats per minute)[13]and the duration of the ventricular action potential is significantly shorter in rodents[14]compared to humans[15].Additionally,cardiomyocytes differ in the proteins expressed in the myofilaments,which affects repolarization and calcium sensitivity[13].One potential strategy to improve the utility of animal models is to create “humanized models” using genetic engineering[11]or engrafting animals with human cells or tissues and immune suppressing them to prevent rejection of the foreign material[16].Although this type of model is useful for studying many conditions including cancer[17],infectious diseases[18]and liver disease[19],they have important limitations,especially in terms of time,cost and difficulties in creation and maintenance.Furthermore,these hybrid animal models are often not feasible for studying the heart and cardiovascular system.
CELLULAR MODELS FOR CARDIOVASCULAR DISEASE
The adult mammalian heart is composed of multiple cell types,including cardiomyocytes,fibroblasts,endothelial cells,vascular and perivascular cells.The composition of the heart varies greatly between species[20]but,in humans,cardiomyocytes are the dominant cell type by volume,encompassing 70%-85% of the total heart.Cardiomyocytes give rise to specialized cells such as atrial myocytes,ventricular myocytes and Purkinje cells[21]and are responsible for the generation of contractile force[22].However,although the other cell types only account for a small portion of the overall total myocardial mass,they are essential for maintaining homeostasis by providing the extracellular matrix and intercellular communication networks necessary to ensure proper cardiac function[23].Although cardiomyocytes may be dominant by volume,they are not the most abundant cells.Fibroblasts are actually the most common cell type in the heart and are vital for maintaining the structure,mechanical and electrical functions of the heart[24].Cardiomyocytes and fibroblasts are the best-studied cardiac cells and,since both cell types have important functions in the heart,we would suggest that both need to be examined to fully comprehend the cardiomyopathies.
Cell culture,using cardiomyocytes,fibroblasts and other cardiac-related cells,represents another well-established system to study human biology,understand disease and assess response to therapeutics.Primary cells and immortalized cell lines derived from human tissues represent two commonly-used experimental models.Primary cells reflect disease biology most faithfully since they are directly isolated from the tissue of interest and they maintain the morphology,function and protein markers in the dish as they possessedin vivo,but they are relatively delicate cells that can be difficult to maintain in culture and have a finite lifespan with limited potential for expansion[25].Immortalized cells are derived by altering cell-cycle check points or modifying telomerase activity and,although these cells don’t have a limited lifespan and are capable of sustained active proliferation,they frequently contain genetic aberrations that can accumulate over time and lead to cellular behaviours that are distinct from those demonstratedin vivo[26].
Another approach to model disease involves the use of patient-derived cells.These cells are obtained from an individual patient and therefore allow for the study of human disease in its original genetic context and also have important advantages over primary or immortalized cells.The two most commonly used patient-derived cell types used for research today are induced pluripotent stem cells (iPSCs) and fibroblasts.Given that the genetic background for an individual is preserved,the use of these patient-specific cells represents perhaps the best tool to realize personalized medicine[27].Personalized medicine refers to a health care approach which recognizes each person’s distinct genetic,clinical and environmental history[28].Personalized medicine ideally adapts therapeutics in order to ensure the best response and safety for the treatment of specific diseases with an individualized approach[29].Using patient-specific cells can help realize this vision by helping researchers identify and understand individual differences.
In conclusion,there are important differences between model systems (Table 1),with advantages and disadvantages that are often dependent on the condition being studied.In reality,a combination of models enabling bothin vivoandin vitrostudies is often required.In this paper,our main focus will be to discuss and compare the different cell types which could be useful for studying genetic cardiomyopathies as an alternative to primary cardiac cells.We will illustrate our discussion with examples of mitochondrial cardiomyopathies that have been studied using different cellular models.
IMMORTALIZED CELL LINES
Immortalized cells are defined as cells whose proliferative capacity has been enhanced using different methods[30].There are a variety of established approaches to immortalize cell lines including the introduction of oncogenes[31-33],viral transformation[34,35],the inactivation of tumor suppressor genes[36,37]or the inactivation of telomere-controlled senescence[38].The establishment of immortalized cell lines has helped the scientific community to study different biological and molecular events[26],although,this approach has been questioned since these immortalized cells differ significantly from cells with an intact cell cycle control and they are more similar to malignant cells in many respects.Therefore,the results obtained with these cells can potentially be misleading if these differences are not considered[39].However,the use of immortalized cells still remains one of the most popular models for the study ofdisease.

Table 1 Comparison between animal and cell models
Immortalized cells have been used to study two inherited diseases caused by point mutations in mitochondrial DNA (mtDNA),mitochondrial myopathy,encephalopathy,lactic acidosis and stroke-like episodes (MELAS),and myoclonic epilepsy and ragged-red fibres (MERRF).In both diseases,an alteration in the posttranscriptional modification of a uridine located in an essential position of specific mitochondrial tRNAs,causes oxidative phosphorylation impairment that leads to the inability to generate sufficient ATP to meet the energy demands of the cell[40].These mitochondrial disorders can be caused by mutations in several genes but,in this example,the immortalized cells were used to model the effect of an A>G transition at nucleotide 3243 in the tRNALeugene causing MELAS[41]and a A>G change in the tRNALysgene at position 8344 causing MERRF[42].Two different studies recapitulated these diseases using cybrid cells[43,44].Cytoplasmic hybrid cells (cybrid) are created using a recipient cell line called rho-zero cells,whose mtDNA has been depleted but the nuclear DNA remains intact and a donor cell which provides mtDNA to the union[45].This approach has the advantage of being able to isolate mtDNA from a donor patient with a specific mtDNA mutation,allowing for the study of the pathology in an immortalized cell line.
Another rare human disorder,Barth syndrome (BTHS) was studied using immortalized cell lines.BTHS is an X-linked recessive disorder characterized by earlyonset cardiomyopathy (usually LVNC or DCM),skeletal muscle weakness and neutropenia related to abnormal mitochondrial structure[46].Disease severity is highly variable,with patients ranging from being asymptomatic to having severe cardiomyopathy and end-stage heart failure[47].Studies have shown that BTHS is caused by loss-of-function mutations in the tafazzin (TAZ) gene[48].TAZis a phospholipid transacylase located in the inner mitochondrial membrane and is responsible for remodeling of the phospholipid cardiolipin[49]which is an essential component of the mitochondrial membrane[50,51].TheTAZgene consists of 11 different exons[52]and mutations have been identified in each exon,primarily missense mutations,although small insertions and deletions have also been found[53].
To study BTHS,the authors used a myoblast cell line (C2C12) derived from mouse skeletal myoblast cells,which is commonly used as a model of disease in mammals for skeletal muscle disorders and myopathies[54,55].The authors designed a stable TAZ knockout (KO) using clustered regularly interspaced short palindromic repeats(CRISPR) technology to target exon 3 in mouse TAZ and cloned it into a plasmid together with the Cas9 nuclease and co-transfected into the cells with a plasmid that allowed for selection with puromycin[56].With the introduction of the plasmids into the cell,the guide RNA binds to exogenous exon 3,and this binding is recognized by the nuclease,which performs the cutting of the gene,disrupting it.The clone whose genomic TAZ DNA band was fragmented into three pieces was the chosen one to be the model of the disease.According to the authors,this model served to recapitulate BTHS,being consistent with other previous models,showing mitochondrial defects such as accumulation of monolyso-cardiolipin,impaired mitochondrial respiration and increased mitochondrial ROS species[56].
Although these studies have used different immortalized cell models,these might not be the best tool to recapitulate the diseases with accuracy.First of all,these cells are derived either from tumors or from the immortalization of other cell times where the cell cycle or the telomerase activity is compromised,therefore,these cells do not resemble normal cell lines in terms of replication and lifespan and,consequently,this can cause genetic and phenotypic variation over time leading to create heterogeneity in the same cell line[57].Secondly,these cell lines,like all cell lines are vulnerable to contamination (e.g.,Mycoplasma) which can remain undetected and modify cell behaviour and gene expression[58].Finally,the use of cellular models generated by using techniques that knockout a gene in particular in a cell line,might not be sufficient to recapitulate the entire spectrum of disease since additional genetic modifiers are not reproduced.
FIBROBLASTS
Fibroblasts are the major stromal cell-type present in connective tissue and are characterized by a flattened and elongated shape with a central nucleus[59](Figure 1).They are derived from mesenchymal precursors and are part of a heterogenous collection of cells widely distributed over the body.Fibroblasts play an important role in connective tissue by producing extracellular matrix compounds,principally collagen type I and III.Fibroblasts not only have a structural role but they are able to repair damaged tissue by migrating to the site of injury and rapidly proliferating to restore the wounded area[60].This proliferation potential explains why fibroblasts are so widely used and why they growin vitrovery easily[61].In addition to their growthrelated properties,fibroblasts are also increasingly recognized as an important contributor to cardiac biology through cell-cell signalling and physical interactions[62,63].Unfortunately,fibroblasts have distinct electrophysiological properties and these cells are not electrically excitable despite the presence of multiple ion channels,including potassium and sodium channels[64].Fibroblasts also lack a specific cell surface marker that distinguishes them from other cell types[65].However,they can be isolated from a skin biopsy and grown in culture[66]but they do have a limited lifespan[67],so their use to study function,structure and disease mechanism is limited to cells that have not undergone an excessive (< 20) number or cell divisions or passages[66].
Fibroblasts have also been used to study MELAS and MERRF.This study demonstrated that the tRNA point mutations did not modify the number of normal mitochondria but there were important differences found regarding the number of secondary lysosomes and residual bodies in both diseases compared to the control cells[68].Furthermore,in both diseases,there was impaired respiratory enzyme activity which decreased mitochondrial respiration rate and membrane potential and impacted cell viability due to the inability to synthesize enough ATP to meet the energy requirements of the cell[68].Even though the cell types affected by MELAS and MERRF in humans are mainly neurons and myocytes[69,70],the easily obtainable skin fibroblasts were sufficient to provide a helpful model to understand some of the mechanisms by which these cell types are compromised.Fibroblasts were also used in BTHS to help understand the molecular basis of the disease.As previously mentioned,diverse mutations have been found in each exon of TAZ,however,there is no clear correlation between the gene mutation type and the different patient phenotypes[71].The authors used fibroblasts from pediatric patients to correlate the severity of the disease with cellular lipid abnormalities and found that there was abnormal composition of cardiolipin,phosphatidyl-choline and phosphatidylethanolamine[72].In this study fibroblasts allowed the distinct lipid composition for each patient to be characterized,which enabled insight into the phenotypic complexity of the disease[72].
Although all these studies successfully used fibroblasts to analyze different mitochondrial cardiomyopathies,all studies had to work within the limitation of fibroblast passage number.The passage number refers to the number of times that the cell can undergo cell division and replication.Studies have shown that,with every passage,the number of mitochondria decreases and that there are changes in the structure of these organelles[73].If not recognized and controlled for,these changes have the potential to mislead researchers into making false conclusions regarding mitochondrial morphology and function.
IPSCS
iPSCs were first created in 2006 after Shinya Yamanaka successfully reprogrammed adult mouse fibroblasts into iPSCs by introducing the pluripotency factors Oct3/4(Octamer binding transcription factor 3/4),Sox2 (sex determining region Y)-box 2),c-Myc and Kfl4 (Kruppel Like Factor-4) under embryonic stem cells (ESC) conditions[74].ESCs are derived from the inner cell mass of mammalian blastocysts and possess selfrenewal capacity,the ability to grow with an unlimited lifespan and the ability to maintain pluripotency and differentiate into every cell type of the three germ layers[75,76].The iPSCs created with these “Yamanaka factors” showed the morphology(Figure 2),proliferative properties and gene expression associated with pluripotency in ESCs[74]but,importantly,did not have to be derived from discarded human embryos.Currently,iPSCs can be created from a variety of mature,differentiated cells most commonly fibroblasts and peripheral blood mononuclear cells[77].
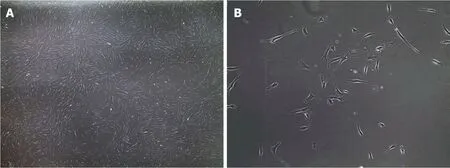
Figure 1 Bright field microscopy images of human fibroblasts.
There are several technical approaches for the delivery of the four critical pluripotency factors necessary for cellular reprogramming to occur[78].There are integrating methods that include retroviral transduction[74],lentiviral delivery[79]and non-integrative methods such as adenoviral transduction[80],plasmid DNA (episomal)transfer[81],lox p lentivirus delivery[82],Sendai virus delivery[83],piggyBAC transposon[84],protein-mediated (polyarginine-tagged polypeptide)[85]and modified synthetic mRNA[86](Table 2).Each methodology has its advantages and disadvantages[87-89]and the choice of delivery vector can have important implications in downstream applications and,therefore,needs to be considered carefully.
Once created,iPSCs have significant advantages compared to other cell types as a model of disease.Since they possess the ability to self-renew,there is no concern about how many passages the cells can tolerate and these cells can be relatively easily expandedin vitroand be used for many experiments[90].Furthermore,since they can be differentiated into mostly every cell type[91],researchers can generate patientdisease- and tissue-specific cells for the disease of interest.
DIFFERENTIATION of IPSCS INTO CARDIOMYOCYTES
Most applications using iPSCs to study human heart disease have differentiated them into beating cardiomyocytes[92]although one group (discussed later) took a rather unique approach and differentiated the iPSCs back into fibroblasts[93].There are several different published and commercial methods to differentiate iPSCs into cardiomyocytes all of which are generally based on the signaling factors that are part of the developmental pathway of cardiomyocytesin vivo[94-96](Figure 3).
Although the ability to generate patient- and disease-specific beating cardiomyocytes is a powerful tool for the study of individual cardiomyopathies[97],the cardiomyocytes that are generated using current methods do have some limitations.First of all,following differentiation,the final population of cardiomyocytes are not completely homogeneous.Differentiated cells contain a mixture of atrial,ventricular and Purkinje cell-types with variable functional properties[98].If a homogeneous population is desired,it may be necessary to select for the cellular subpopulation of interest using sorting techniques based on surface marker expression[99]or genetic selection[100]which further complicates the process requiring additional time and expense and exposes the cells to additional handling and stresses which they may not survive.Furthermore,for some cell types,e.g.ventricular myocytes,unique cell surface markers do not exist[101].Another issue is that the cardiomyocytes obtained using current differentiation strategies have a phenotype resembling fetal cells in terms of structure,molecular markers and metabolism[102].This lack of maturity can require additional steps (which are not fully established or reliably reproducible at this time) or additional time in culture to obtain a more adult-like cardiomyocyte population[103]Several methods to stimulate the maturation of iPSC-derived cardiomyocytes have been published based upon electrical[104],mechanical[105],chemical stimulation[106]or matrix modification[107].This is currently an area of active investigation and future advances and improvements are certain which will further enhance the utility of iPSC-CMs for the study of genetic cardiomyopathies.However,even with these functional limitations of derived cells,they have been helpful for scientists seeking insight into cardiac biology and disease[108,109].
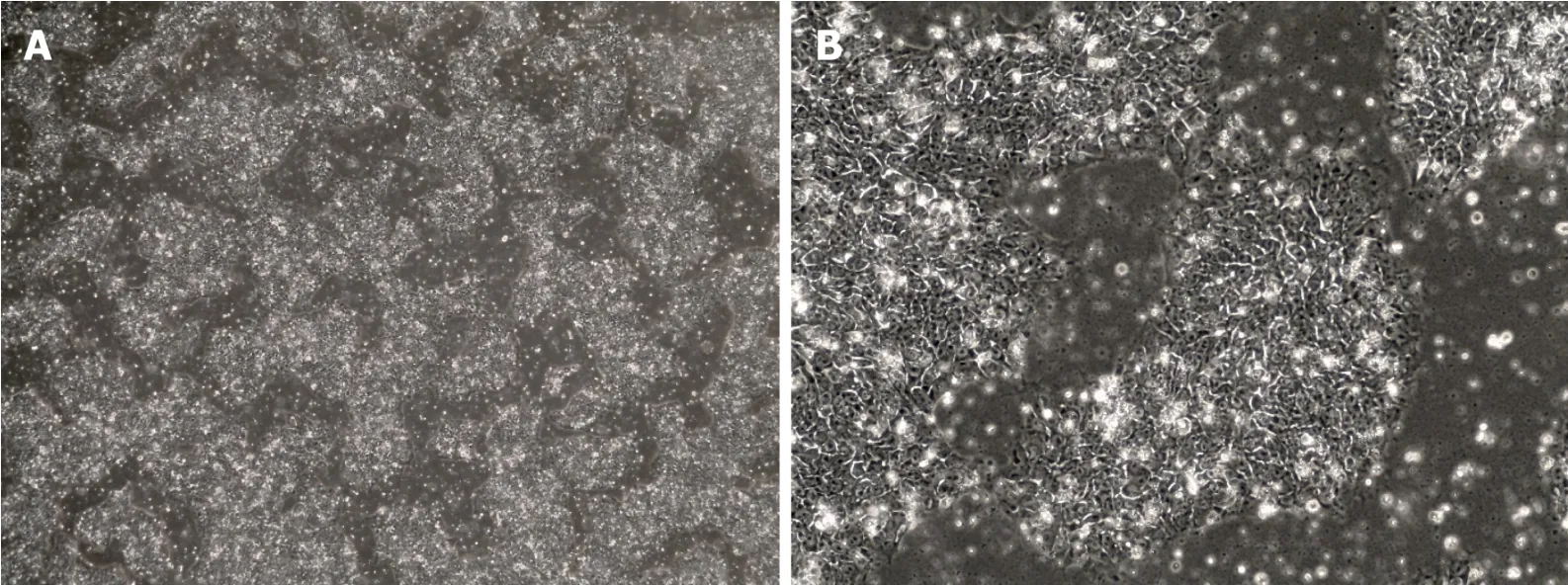
Figure 2 Bright field microscopy images of human induced pluripotent stem cells.
STUDYING GENETIC CARDIOMYOPATHIES USING IPSCS
Primary fibroblasts from a patient with MELAS were reprogrammed into iPSCs using a retroviral approach in order to establish a novel disease model[110].As standard practice,the differentiation capacities of the iPSCs were tested using a teratoma formation assay to demonstrate that the cells were capable of generating all germ layers and immunocytochemistry for the pluripotency markers Oct-4 and SSEA-4 was performed to confirm pluripotency.Tissues in MELAS patients can vary in the levels of abnormal mitochondria (heteroplasmy)[111]so the researchers assessed this in patient cells using quantitative real-time PCR to measure mutation ratios and mtDNA copy number.They found that different fibroblast lines had different levels of heteroplasmy ranging from < 5% to 95%.They then demonstrated that those fibroblasts with lower levels of heteroplasmy showed increased heteroplasmy after several passages while those with higher levels did not vary significantly after multiple passages.There were also variations with regards to mtDNA copy number after each passage.This data suggests that the mitochondrial abnormalities in patient fibroblasts can change over time in culture.However,because of their importance in cardiac biology,the authors still wanted to study MELAS.Therefore,the MELAS iPSCs were differentiated back into fibroblasts but,because of the unique selfrenewing properties of iPSCs,the authors could overcome passage-associated changes in the mitochondria.In the fibroblasts derived from patient iPSCs,levels of heteroplasmy were found to be similar to the iPSCs from which they were differentiated.These iPSC-derived fibroblasts were then characterized with regards to the enzymatic activities of the mitochondrial respiratory complexes and compared to primary skin fibroblasts.These studies revealed that the iPSC-derived cells recapitulated the disease phenotype and did not demonstrate altered levels of heteroplasmy in culture and therefore represent a unique and novelin vitromodel of MELAS[110].
MERRF has also been studied using retrovirus-reprogrammed iPSCs.In this study,they generated iPSCs from patient dermal fibroblasts.After reprogramming the fibroblasts using OCT4,SOX2,KLF4,and GLIS1 delivered into the cells,they differentiated the resulting iPSCs into the two different cell types most involved in the disease,cardiomyocytes (iPSC-CMs)[112]and neural progenitor cells (iPSC-NPCs).When they tested all three cell types,they found that all MERRF patient-derived cells(iPSCs,iPSC-CMs and iPSC-NPCs) had reduced oxygen consumption,elevated reactive oxygen species (ROS),reduced growth and fragmented mitochondria.The cellular phenotype correlated with the molecular mechanism of the disease,allowing iPSCs and iPSC-derived cells to serve as a model for the disease[93].
Differentiated iPSCs have also been used in the study of BTHS.The cells of two unrelated patients were reprogrammed using either retroviral[113]or modified RNA approaches[114].These two patients had different mutations in TAZ,one having a frameshift mutation and the other a missense mutation.After the generation of the iPSCs,they differentiated them into cardiomyocytes that they then used to create tissue layers and a heart-on-chip model[115].The iPSC-CMs showed abnormalities in cardiolipin processing,sarcomere assembly,myocardial contraction,ROS production and cardiomyocyte functioning,correlating with the abnormalities and cardiacdysfunction observed in patients,demonstrating again that is possible to use anin vitromodel to provide insight into human disease and test potential therapeutics[116].
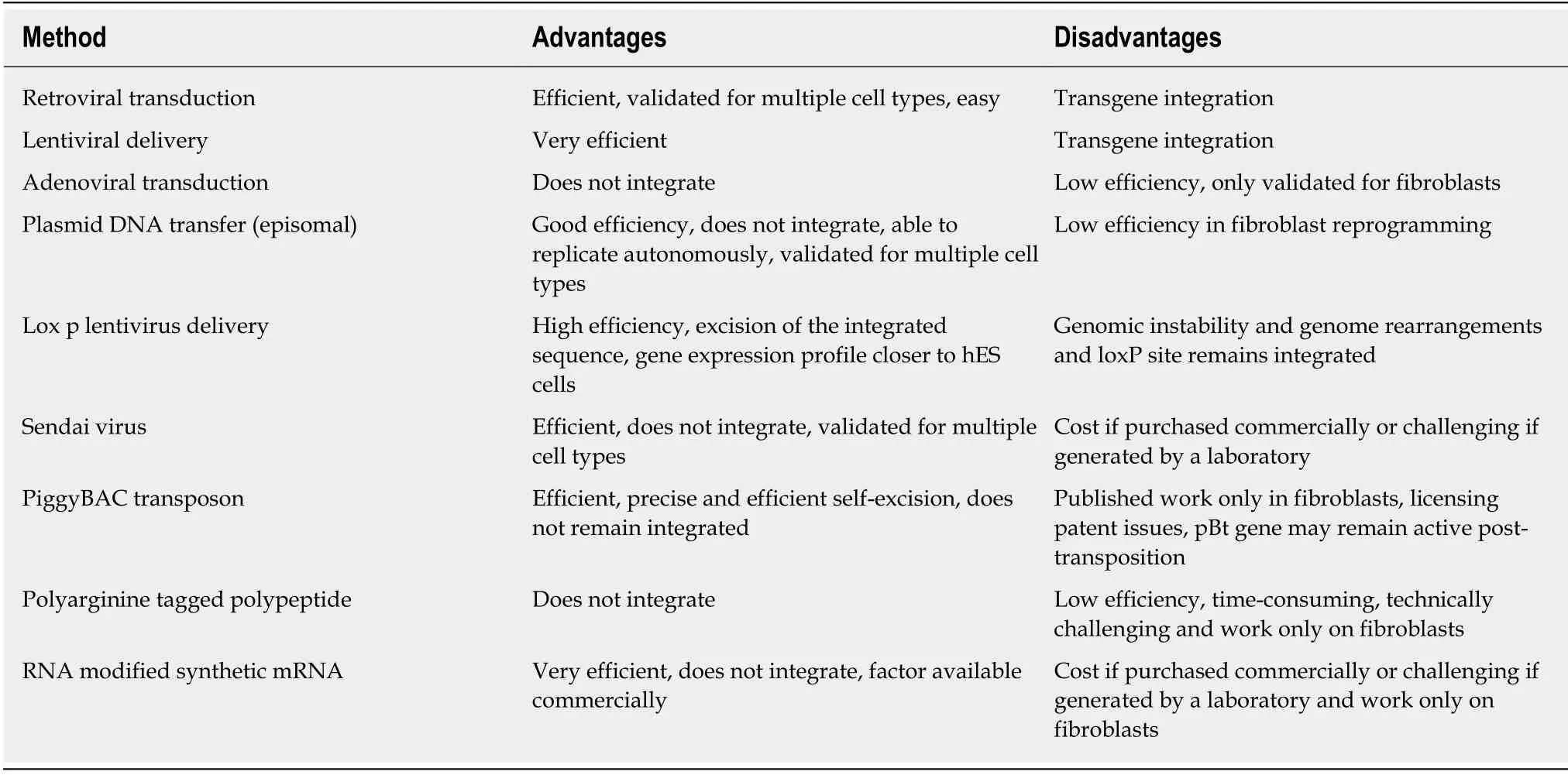
Table 2 Methods of delivery for reprogramming factors
iPSC-CMs have also been used to study other cardiomyopathies.For example,iPSC-CMs have also been used to understand the pathological effects caused by the reduced expression of frataxin (FXN) in Friedreich ataxia (FA).This neurodegenerative disease is caused by the expansion of a short tandem repeat (GAA) in theFXNgene,which can result in transcriptional silencing[117]and therefore,the development of HCM[118]which is an important component of the disease phenotype but its development is not understood.In this study,the researchers generated iPSCs from three patients using an episomal reprogramming approach and then differentiated the resulting iPSCs into cardiomyocytes[119].Analysis of the iPSC-CMs showed that these cells had an increased beating rate which was related to a defect in calcium handling.Therefore,these cells revealed novel biology that could potentially contribute to the future development of treatment for this disease[120].It is important to note that this cellular phenotype could arguably not have been accomplished with any other cell type.
The DCM with ataxia syndrome (DCMA) is an autosomal recessive disorder caused by mutation in DNAJC19 and is characterized by 39% mortality[121]during early childhood due to severe heart failure[122].DCMA has been related to BTHS due to the presence of metabolic abnormalities (i.e.production of 3-methylglutaconic acid) and abnormal mitochondria are thought to be responsible for heart failure[123].Rohaniet al[123]successfully established four patient iPSC lines that have been differentiated into CMs expressing cardiac-specific markers and this will allow for the study of four unique patient cell lines.This disease still needs to be further characterized but the use of iPSC-CMs derived from patients looks promising as a cellular model to provide a better understanding of the disease.
Finally,iPSC-CMs have also been used to study familial HCM,characterized by thickened left ventricular walls,myofiber disarrays and myocardial fibrosis that often results in arrhythmias[124].This can be caused by different mutation in at least 11 different genes which encode sarcomeric proteins[125].In this study,the authors generated iPSC-CMs derived from an HCM patient that had a single missense mutation in the β-myosin heavy chain (MYH7) gene.Whole transcriptional analysis of these iPSC-CMs provided useful insights into the disease,revealing important signaling pathways implicated in the pathogenicity of HCM[126].
FIBROBLASTS VS IPSCS
As we have described,both fibroblasts and iPSCs have been used to model genetic cardiomyopathies and both cell types have important advantages and disadvantages(Table 3).The characteristics of a specific cell type and the disease being studied may have an important influence on the researcher’s choice of cellular model and,in some situations,the study of both fibroblasts and iPSCs may be complementary.For instance,in a disease in which the interaction between cardiomyocytes and fibroblast plays a role in the development of the pathogenesis,for example in cardiac fibrosis and arrhythmias[127],the study of both cell types would likely be beneficial.
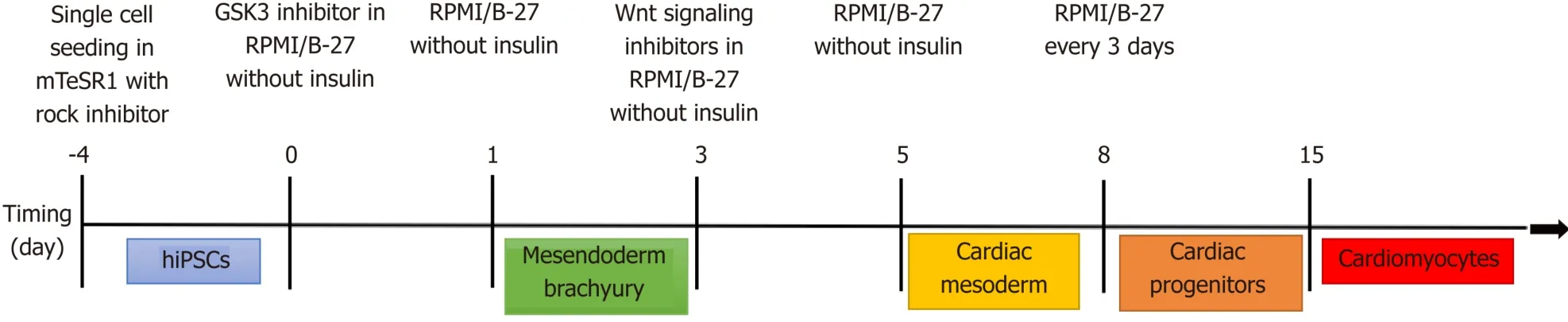
Figure 3 Cardiomyocyte differentiation protocol.
In order to solve the lifespan problem with primary cells such as fibroblasts,reversible immortalization could be performed to increase the number of passages and limit the risk for the development of aberrations in the genome[128].In one study,this reversible immortalization was performed in primary neonatal rat cardiomyocytes using lentiviral transduction with either simian virus 40 large T antigen(TAg) or Bmi-1 together with the human telomerase reverse transcriptase (hTERT).After the cells were expanded,the introduced genes were removed using an adenoviral vector expressing Cre recombinase.The transduction of Bm1-1/hTERT into the primary cardiomyocytes successfully immortalized the cells and they maintained the expected cell morphology and presence of contact inhibition,suggesting that the cells had not become aberrant during the immortalization process[129].This technique is an example of how genetic engineering could be used to overcome some of the limitations of cell biology which may be useful to researchers seeking to study a particular cell type.
Although patient-derived iPSCs and the differentiated cells that are created are excellent models of disease,the generation of appropriate controls is essential since they will help to define the abnormal phenotype.For some diseases that are enriched in specific populations with a unique genetic background,for example,DCMA,which is highly prevalent in the Hutterite population of southern Alberta[130],there is a need for controls who also have the same genetic background.The Hutterites are an isolated and genetically-closed population descended from a limited number of European ancestors with a communal religious lifestyle[131].CRISPR/Cas9[132]can be used to repair the DNA mutation in patient cells to create isogenic controls[133]that are genetically identical except for a single genetic mutation background[134].
CONCLUSION
Cellular models represent an important tool for investigating rare human diseases including the genetic cardiomyopathies.Generic immortalized cells are the most commonly used cell model as they are the easiest to handle in terms of proliferation capacity,growth rate and low maintenance and can be easily genetically manipulated.Conversely,obtaining cells from individual patients allows the study of interindividual differences and the important role of genetic modifiers in shaping disease phenotype and increases the possibility of developing personalized therapeutics.Certainly,in vitromodels have some significant limitations but,in many cases,can provide a model that is otherwise not available.Particularly for cells differentiated from iPSCs,it is true that further research is necessary to optimize these cells but the potential for the development of an accurate and personalized cellular model is very promising for those diseases where conventional cells and animal models are limited.
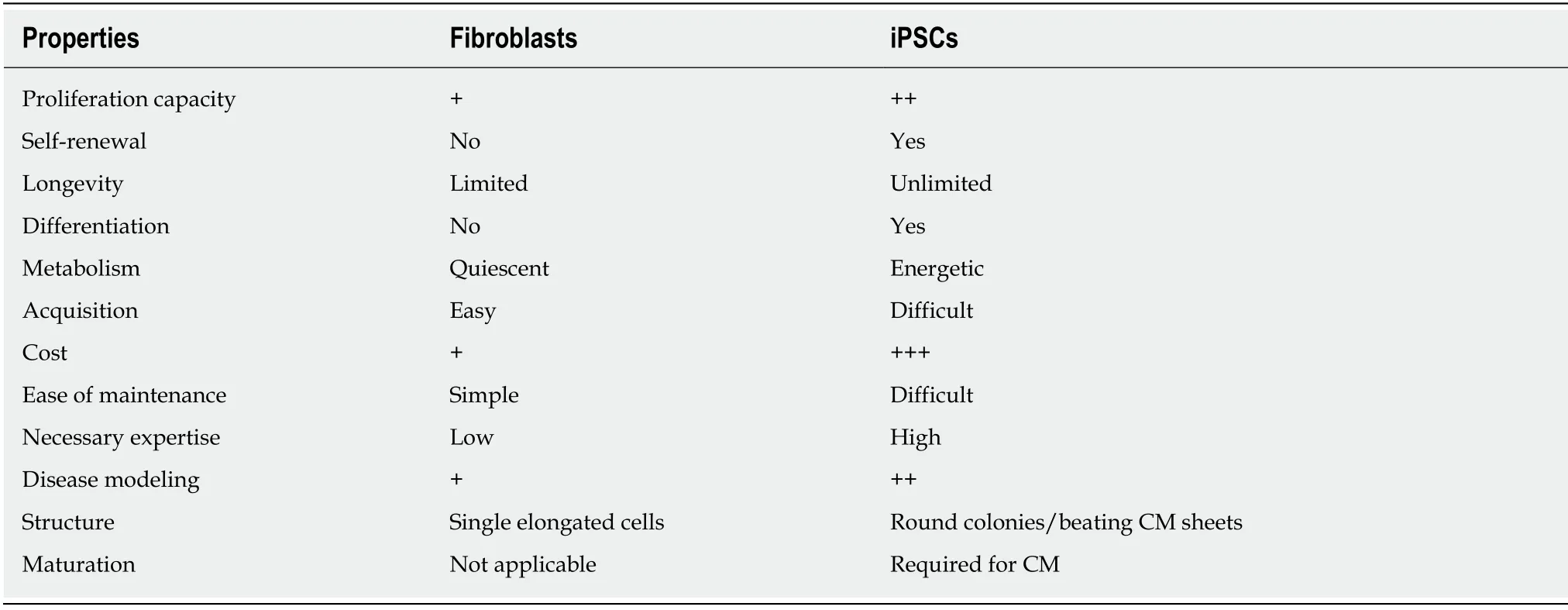
Table 3 Advantages and disadvantages of different cell types for modeling disease in vitro
