1例儿童自身免疫内分泌腺病综合征Ⅲ型诊治分析的报道
2019-10-30符芸瑜杨锐
符芸瑜 杨锐


[摘要]儿童自身免疫内分泌腺病综合征在临床上较为少见,一旦出现,容易误诊、漏诊,如不及时明确诊断、及时治疗,往往会对整个疾病的诊疗过程造成一定延误。本文对2016年9月入院的1例儿童自身免疫内分泌腺病综合征Ⅲ型患者进行分析与总结,探讨自身免疫内分泌腺病综合征的诊断、治疗,结合实际的发病及诊疗过程,讨论如何对该病作出诊断、分型及相应的治疗;提出该病需要长达数十年的随访,可能出现分型的改变,以便及时给予相应的诊断及治疗措施。
[关键词]自身免疫腺病;1型糖尿病;儿童;Graves病;综合征
[中图分类号] R593.2 [文献标识码] A [文章编号] 1674-4721(2019)8(c)-0192-03
[Abstract] Children autoimmune polyendocrinopthy syndrome was rarely seen in the clinic, once it appeared, it might be easily missed and misdiagnosed, if not diagnosed and treated in time, the whole process of diagnosis and treatment will be delayed. One case of children autoimmune polyendocrinopthy syndrome type Ⅲ admitted in September 2016 was analyzed and summarized in this article, the objective was to probe into the diagnosis and treatment of the autoimmune endocrine adenopathy syndrome, combining with the actual paroxysm and treatment course, the diagnosis, classification and relevant treatment were discussed. This article also proposed that the disease needed follow-up for more than ten years, and there might exist the possibility of classification changes, in order that corresponding diagnosis and treatment measures could be given in time.
[Key words] Autoimmune adenosis; Type 1 diabetes mellitus; Children; Graves disease; Syndrome
自身免疫内分泌腺病综合征(autoimmune polyendocrinopthy syndrome,APS)是临床上的罕见病,儿童发病、诊断更是少之甚少。临床上大多患者首诊见于其他专科,随着该病患病率的增高及对疾病认识水平的提高,最终常由内分泌专科确诊、治疗。APS是指由于自身免疫因素引起或导致2个或2个以上内分泌腺体功能异常的综合征,除此之外,该病还可以累及其他非内分泌系统,如消化系统、血液系统、神经系统等,大多数病变表现为腺体功能低下,也可见功能亢进;各腺体的病变可以同时发生,也可以相继发生,甚至相差数十年发生。现报道1例儿童自身免疫内分泌腺病综合征Ⅲ型病例的诊治分析。
1 病例资料
患儿男,11岁,因“口干、多饮、多尿、消瘦1个月”于2016年9月来院就诊,1个月来体重减轻6.5 kg,查空腹指尖血糖>33.3 mmol/L,诊断为“糖尿病”收入院治疗。既往病史:患者于2016年5月起出现消瘦,颈前肿物,于南方医科大学珠江医院就诊,检测甲状腺功能提示:游离三碘甲状腺氨基酸:36.06 pmol/L,游离甲状腺素:64.63 pmol/L,促甲状腺激素:0.01 mIU/L,促甲状腺素受体抗体:13.2 IU/L,抗甲状腺球蛋白抗体:13.91 kU/L,甲状腺过氧化物酶抗体:117.5 kU/L。診断“甲状腺功能亢进症”,予甲巯咪唑100 mg/d。此后每月定期复查甲功、血常规、肝功能,抗甲亢治疗期间血常规及肝功能未见明显异常,甲状腺功能指标逐渐改善。既往无其他特殊病史。否认类似家族遗传病史。
入院查体:体温(T)36.2℃,脉搏(P)116次/min,呼吸(R)20次/min,血压(BP)123/66 mmHg。发育正常,营养中等,形体消瘦,皮肤、口唇干燥,无黑棘皮、痤疮、紫纹,全身浅表淋巴结未触及肿大。双眼眼球突出,眼裂增宽,Joffroy征(-)、vonGreafe征(-)、Stellwag征(-)、Mobius征(-),球结膜无充血和水肿,巩膜无黄染。双侧甲状腺Ⅲ度肿大,无压痛,未闻及收缩期动脉杂音。呼吸略促,双肺部呼吸音粗,未闻及干湿性啰音。叩诊心界无扩大,心率(HR) 116次/min,律齐,各瓣膜听诊区未闻及病理性杂音。腹软,无压痛及反跳痛,肝、脾肋下未触及,肾区无叩击痛。双下肢无水肿。双股动脉、腘动脉、背动脉波动正常,四肢浅感觉正常。余无特殊。
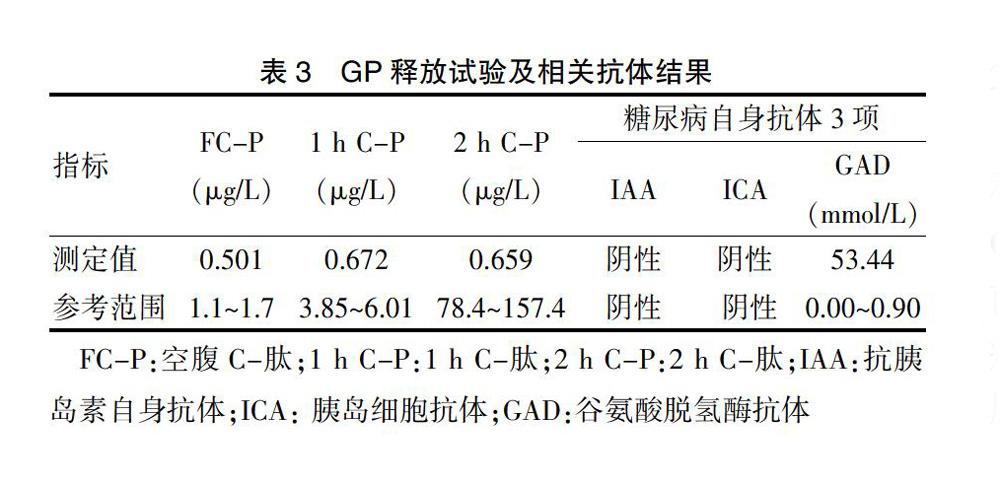
实验室检查结果见表1,表中未列出的血常规、肝功能、肾功、电解质均正常。同时行多内分泌腺体功能检查,结果见表2~4,其中未列出的24 h尿游离皮质醇、甲状旁腺激素、孕酮、泌乳素、促卵泡雌激素、促黄体生成素均正常。免疫系统检查:风湿指标:抗链球菌溶血素“O”(ASO):270 kU/L(0~100 kU/L),类风湿因子(RF)正常。抗核抗体谱、血管炎指标八项、抗中性粒细胞胞浆抗体、抗磷脂综合征指标均正常,该患者所有检验、检查结果均出自南方医科大学珠江医院。
2 治疗及转归
患儿为男性儿童,此次因糖尿病入院,起病急、病程短,自发酮体倾向,C-P水平差,糖尿病抗体3项中GAD阳性,明确1型糖尿病。患者在发生1型糖尿病前出现甲状腺功能亢进症,进一步检查明确Graves病;根据完善的相关实验室检查结果可暂时除外肾上腺皮质病变以及其他免疫系统疾病,综合考虑该患者为自身免疫多发内分泌腺病综合征APSⅢ型:T1DM,Graves病;1型糖尿病 糖尿病酮症。给予小剂量胰岛素静脉点滴、补液消酮、纠正电解质紊乱、改善循环和对症治疗,入院第2天复查酮体消除后给予胰岛素泵皮下泵入控制血糖、补液、纠正电解质紊乱、抗甲亢、改善循环和对症治疗,血糖逐渐平稳,口干、多饮、多尿、乏力症状消失。后改为门冬胰岛素30笔芯皮下注射联合午餐时口服二甲双胍片0.5 g控制血糖,根据血糖波动情况调整胰岛素剂量。全天血糖波动:4.6~18 mmol/L。入院后继续口服甲巯咪唑10 mg,2次/d。
经治疗12 d后病情稳定出院,出院时全天血糖波动:3.5~11.3 mmol/L。1个月后随访,患者无口干、多饮、多尿、乏力症状,体重增加3.5 kg,血糖较前下降、平稳,波动于4.5~10.6 mmol/L。嘱其继续长期应用胰岛素,暂时联合二甲双胍片控制血糖,继续口服甲巯咪唑抗甲亢治疗,定期复查血糖、甲状腺功能,定期随诊。
3 讨论
APS是指2个或2个以上的内分泌腺体因自身免疫功能缺陷而发生功能受损为主要表现的一系列综合征,可以累及非内分泌腺器官[1]。APS患者表现为多器官功能障碍相互影响,其中多数表现为器官(或细胞)功能减退或衰竭,个别器官(或细胞)表现为功能亢进。根据各个器官受累情况,APS既往分型为4型,具体分型如下。
3.1 APSⅠ型
这一型以往也称为黏膜皮肤念珠菌内分泌病。主要有以下4种和(或)病变,肾上腺皮质功能减退(AI)或肾上腺皮质自身抗体阳性﹑甲状旁腺功能减低(HP)﹑慢性皮肤黏膜念珠菌感染三者中至少两种病变。有些患者仅有其中一种内分泌病变,或者四种均有,但这四种病变起病时间各可相距十到数十年。除此之外,该型的患者还可以有垂体炎﹑多囊卵巢﹑秃发﹑白癜风﹑活动性肝炎,罕见的有类风湿关节炎﹑干燥综合征[2-4,7]。
3.2 APSⅡ型
本型是AI并自身免疫性甲状腺疾病(AITD)和(或)1型糖尿病[5]。该型不伴有黏膜念珠菌感染[6-7]。也有报道此型可能合并生长激素缺乏[8-9]。
3.3 APSⅢ型
APSⅢ型是指AITD伴有一个或多个自身免疫性疾病,但是不伴有Addison病和(或)HP。除此之外,该型的患者还可以合并性腺功能减退、淋巴细胞性垂体炎、胰岛素自身免疫综合征(Hirata病)、慢性萎缩性胃炎、恶性贫血、自身免疫性皮肤及神经肌肉疾病、系统性自身免疫性疾病等[10-11]。
3.4 APSⅣ型
这一分型目前最少见,是指两种或两种以上内分泌腺(器官)发生自身免疫性疾病,为AI合并其他自身免疫性疾病,但有异于Ⅰ、Ⅱ、Ⅲ型[12-16],可能合并类风湿性关节炎、原发性肝功能衰竭、乳糜泻等,但不合并念珠菌病、AITD、T1DM。
由于APSⅠ、Ⅱ、Ⅲ型的临床表现常有重叠,因此,近年有新的分型趋向将这三型合并为一型,单纯将APS分为两型。
综上所述,本例患儿首先发生AITD,继而发生T1DM,体内存在GAD、TRAb、TPOAb-R等多腺体自身免疫证据,无皮肤黏膜念珠菌感染及甲状旁腺功能减退,故不属于APSⅠ型;因未出现AI,故不属于APSⅡ型;综上所述,属于APSⅢ型。由于APS临床表现多样,各种内分泌腺体功能受损的临床症状可以同步也可以不同步发生,容易漏诊误诊。因为各个器官病变发病时间相隔可长达20年,该患者在长期的随访中可能出现新的疾病,导致分型改变,故应接受长期随访。
[参考文献]
[1]陈灏珠,廖履坦,杨秉辉.实用内科学[M].12版.北京:人民卫生出版社,2005:1311-1313.
[2]颜纯,王慕逖.小儿内分泌学[M].2版.北京:人民卫生出版社,2006:626.
[3]Fischer A,Provot J,Jais JP,等.原发性免疫缺陷病易发生自身免疫病和炎症表现[J].中华临床免疫和变态反应杂志,2018,12(2):230-235.
[4]向茜,孙健玮,马琼麟.自身免疫性多内分泌腺病综合征Ⅰ型1例[J].中华骨质疏松和骨矿盐疾病杂志,2018,11(3):284.
[5]孙永香,何亚非,栗夏连.1例自身免疫性多内分泌腺病综合征Ⅰ型患者的临床及家系AIRE基因突变分析[J].中国当代儿科杂志,2016,18(2):147-151.
[6]李杨,黄朱亮,万菁菁.自身免疫性多发内分泌腺病综合征Ⅰ型1例报道[J].国际内分泌代谢杂志,2018,38(1):63.
[7]李杨,万菁菁,李零燕,等.自身免疫性多发内分泌腺病综合征Ⅰ型研究进展[J].国际内分泌代谢杂志2017,37(6):426-429.
[8]Papathanasiou A,Kousta E,Skarpa V,et al.Growth hormone deficiency in a patient with autoimmune polyendocrinopathy type 2[J].Hormones,2007,6(3):247-250.
[9]謝小超,杨爱红,刘敏,等.自身免疫性多内分泌腺病综合征Ⅲ型1例报告[J].中国实用医药,2016,11(24):216-217.
[10]吴昊,李金慧,关海霞,等.自身免疫性多内分泌腺综合征4例报告并文献复习[J].国际内分泌代谢杂志,2016, 36(6):420-423.
[11]赵艳艳,秦贵军.1型糖尿病与其他内分泌疾病[J].中华糖尿病杂志,2016,8(10):583-587.
[12]陈丹霞,麦鸿成,郭叶群,等.自身免疫性多内分泌腺病综合征Ⅲ型合并胸腺增生1例报告并文献复习[J].南昌大学学报(医学版),2017,57(3)98-101.
[13]皮亚雷,张亚男,韩笑,等.罕见基因突变致Ⅰ型自身免疫性多内分泌腺病综合征1例临床及家系分析[J].临床荟萃,2016,31(12):1318-1320.
[14]狄红杰,陈国芳,刘超.自身免疫性多发性内分泌腺病综合征病例报道及分子遗传学研究进展[J].中国实用内科杂志,2018,38(10):26-29.
[15]Barker JM. Clinical.Type I diabetes associated autoimmunity:natural history,genetic associations,and screening[J].J Clin Endocrinol Metab,2006,91(4):1210-1217.
[16]李俊岩,李剑波.自身免疫性多内分泌腺病综合征二例[J].中华糖尿病杂志,2017,9(9):581-583.
(收稿日期:2019-02-18 本文编辑:许俊琴)
