原位氮掺杂大孔径介孔碳材料的制备及表征
2019-10-19潘红艳杨春亮贾双珠许值铭王贤书史永永
王 梦,潘红艳,杨春亮,贾双珠,许值铭,王贤书,史永永,林 倩
(1.贵州大学化学与化工学院,贵阳 550025;2.贵州省绿色化工与清洁能源技术重点实验室,贵阳 550025)
1 引 言
大孔径介孔碳材料,因其具有高的比表面积,大的孔径、孔容,高的机械稳定性和化学惰性等独特的性能,使其在催化[1]、吸附分离[2]、药物载体[3]、电极材料[4]等领域具有广阔的应用前景。
壳聚糖是一种生物质多糖,具有较高的C(43.12wt%)和N(7.689wt%)含量[5],具有良好的生物官能性和相容性、血液相容性、安全性和微生物降解性等性能,因此可作为碳源制备介孔碳。
目前,以壳聚糖为碳源制备介孔碳主要为硬模板法和软模板法。硬模板法通常以SiO2为模板,得到孔径可调的介孔碳。例如,Andrzej等[6]以壳聚糖为碳源,预先制备的胶态SiO2为硬模板,制得孔径在21.6 nm、孔容为4.08 cm3/g的含氮介孔碳。但该方法为两步合成法,步骤繁琐。杨春亮等[7]以壳聚糖为碳源,正硅酸乙酯(TEOS)为硅模板一步法制备出平均孔径为5.3 nm、孔容为1.48 cm3/g的介孔碳。与硬模板相比,软模板法制备介孔碳,具有步骤简便、成本较低、形貌和孔结构更易调节等优点,近年来成为研究的热点。软模板法通常以F127、P123等表面活性剂为模板剂制得介孔碳。例如,冯妙娜等[8]以壳聚糖为碳源,F127为软模板,通过一步碳化,制备出平均孔径在5.276 nm、孔容仅为0.608 cm3/g的介孔碳。Sun等[9]以壳聚糖质子盐为碳源,F127为模板剂,制备得到了孔容仅为0.31 cm3/g的介孔碳。
前人虽然以壳聚糖为碳源、F127为模板剂,采用软模板法制得介孔碳,但其孔容和孔径都较小,从而限制其在生物大分子(如DNA和蛋白质)的吸附与传输[10]、催化剂纳米颗粒(如Ag、Pd)的负载[11]。
要想将以壳聚糖为碳源制备的介孔碳应用在生物大分子方面,必须增大其介孔孔径和孔容。而孔径大小受模板剂尺寸的影响。前人研究指出,F127在水相溶液中会形成疏水段向内,亲水段向外的胶束模板。TEOS在溶液中水解缩聚生成的硅酸聚集体。通过分析发现,硅酸聚集体表面的硅羟基可以与F127亲水段上的醚键发生氢键作用,从而包裹在F127胶束上。F127和TEOS的共模板作用可实现增加模板剂尺寸的作用,从而实现提高介孔碳孔径的目的。当壳聚糖作为碳源时,壳聚糖上的羟基会以共价键的方式与共模板上的硅羟基脱水交联[12],使其包裹在硅-F127共模板外面。通过调节碳源与共模板剂间的比值和其他反应条件,可得到大孔径大孔容的介孔碳。
鉴于此,本文运用软模板机理,以壳聚糖为碳氮源、F127和TEOS为共模板剂,利用混合模板法,一步法制备原位氮掺杂大孔径介孔碳。并考察了硅碳比、pH值、反应温度对超大孔径介孔碳孔结构的影响。使用N2吸附/脱附等温线、FT-IR、XRD、TEM、SEM和XPS对所得介孔碳进行表征分析,本文还研究了介孔碳对牛血清白蛋白(BSA)的吸附能力。
2 实 验
2.1 原料与试剂
壳聚糖(脱乙酰度80.0%~95.0%)从国药集团购买;三嵌段共聚物F127(分子量为12600,分子式PEO106PPO70PEO106)和牛血清白蛋白(BSA)从sigma-aldrich公司购买;试剂级硅酸四乙酯(aladdin,分子量为208.33,分子式C8H20O4Si)从aladdin购买;浓HCl(质量分数37%)和无水乙醇从天津市富宇精细化工试剂厂购买;NaOH从天津市永大化学试剂厂购买;所用水为实验室自制蒸馏水。
2.2 样品的制备
原位氮掺杂介孔碳的制备:将1 g壳聚糖溶解到33 mL含一定量的2 mol/L HCL(2~2.5 mL)的水溶液中,使其完全溶解,得到溶液A;将一定量的正硅酸乙酯(3.7~5.5 mL)逐滴滴入5 mL含有1 g F127和1 mL 0.2 mol/L HCL的乙醇溶液中,继续搅拌10 min,得到溶液B;A和B混合均匀,继续搅拌80 min后,移入培养皿,经过48 h的常温干燥和12 h的80 ℃固化后得到复合物Chitoan-silica-F127,将其移入管式炉中,在氮气保护下,从室温以2 ℃/min升温至410 ℃并保温2 h后,再以5 ℃/min升温至900 ℃并保温2 h,碳化得到碳硅复合物LPMC-X-Y-Z*,将其浸入2~3 mol/L氢氧化钠溶液中,放入85 ℃恒温水浴锅里保温18 h,再经过滤洗涤干燥,得到介孔碳材料。在本文中,以壳聚糖作为碳源和氮源,三嵌段共聚物F127和正硅酸乙酯分别作为软模板和硅模板。考察了硅碳比、pH值和反应温度对介孔碳孔结构的影响。制备的介孔碳材料命名为LPMC-X-Y-Z,其中X、Y、Z分别代表硅碳比、pH值和反应温度。
2.3 样品的结构表征
使用ASAP2020型(美国Micromeritics公司)物理吸附仪测量样品的N2吸附/脱附等温线,样品预先在200 ℃真空脱气6 h,使用BET法在P/P0为0.05~0.3内计算SBET,分别采用H-K方程和BJH方程计算微孔孔容(Vmic)和中孔孔容(Vmes),采用DFT计算全孔孔径分布,在P/P0为0.995计算总孔容(Vtotal);使用Bruker D8 X射线衍射仪对样品进行X射线衍射分析;使用Tecnai G2 F20 S-双透射电子显微镜(TEM)在200 kW工作电压下观察了样品的孔道。使用S-4800 Co,Jpn扫描电镜(SEM)观察了样品的形貌。使用Escalab 250XI光电子能谱仪(美国Thermo Scienftic公司)分析样品元素形态。
2.4 样品的吸附性能评价
测定吸附等温线:将一定量的碳材料LPMC-1.33-4.5-30样品分散于100 mL一定浓度BSA的CH3COOH/CH3COONa缓冲液中(50~1000 mg/L)。在37 ℃、振荡频率80 r/min条件下,恒温振荡24 h。用分光光度计在波长280 nm处测定各溶液中残余BSA的浓度,样品对BSA的平衡吸附量qe按下式计算:
qe=V(C0-Ce)/m
(1)
式中,V是BSA溶液的体积(L),m是碳材料样品的质量(g),C0是BSA溶液的初始浓度(mg/L),Ce是碳材料样品对BSA溶液的吸附平衡浓度(mg/L)。
测定吸附速率曲线:准确称取碳材料LPMC-1.33-4.5-30样品100 mg,置于250 mL具塞锥形瓶中,加入200 mL 浓度为800 mg/L 的BSA水溶液。37 ℃、振荡频率 80 r/min条件下恒温振荡,在不同振荡时间t取上层清液测BSA的浓度,按(1)式计算对应振荡时间点的平衡吸附量,记为qt。
3 结果与讨论
3.1 介孔碳孔结构表征
图1分别是不同硅碳比(a, b)、pH值(c, d)及反应温度(e, f)制备碳材料的N2吸附/脱附等温线及孔结构分布图。由图可见,所有的碳材料在低相对压力时(P/P0<0.1)对N2都有较低的吸附量,表明含有极少量的微孔;然而,除LPMC-1.33-4.5-20外,其余碳材料在P/P0>0.8后,N2吸附量急剧增加,且出现明显的滞后回环,为大中孔的冷凝,说明该类样品含有大量的大中孔。碳材料的图1(b, d, f)的DFT孔径分布进一步证实除LPMC-1.33-4.5-20外,其余碳材料均在<2 nm有极少的微孔分布和大量的大介孔,孔径主要集中在20 nm左右。
表1是所得介孔碳材料的BET表面积、孔体积和平均孔径。可以看出,Si/C比、pH和反应温度对中孔体积和孔径分布都有显著的影响。
由图1(b)和表1可见,随着Si/C比从1.0增加到1.5,所制备介孔碳材料的孔径和介孔孔容先增大后又缩小,在Si/C比为1.33时,样品LPMC-1.33-5.0-40有大的孔径(19.2 nm)和介孔孔容(2.20 cm3/g)。这是因为,随着Si/C比的增加,TEOS浓度增加,在这种溶液中可以生成更多的由TEOS水解缩聚得到的胶体粒子、非线性或网络胶体聚合物。这时,这些胶体粒子的碰撞几率就会增加,在这种溶液中可以形成更大尺寸的胶粒[13]。移除硅模板后形成的碳材料孔径和孔容增大。但是,当TEOS浓度过高时,胶体颗粒的尺寸过大,导致一些较大的胶体粒子在溶液中发生重力沉降,溶胶体系的稳定性降低,壳聚糖不能很好地包裹在这些胶体粒子的外面,使得在较高的Si/C比(1.5)下制备的LPMC-1.5-5.0-40碳材料的平均孔径和介孔体积较小。
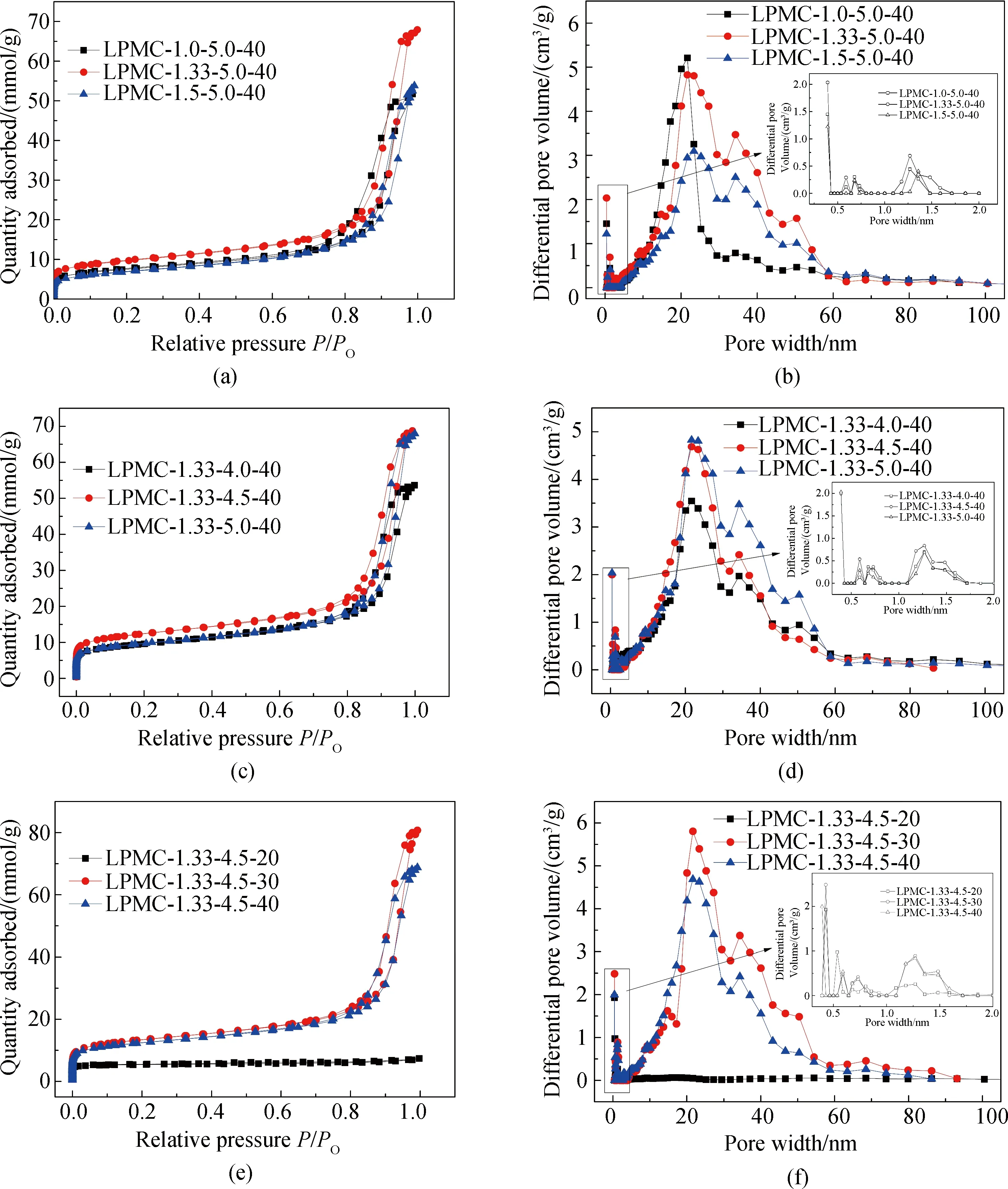
图1 不同硅碳比(a,b)、pH值(c,d)和反应温度(e,f)制备的碳材料的N2吸附/脱附等温线和孔径分布Fig.1 N2adsorption/desorption isotherms and pore size distributions of obtained carbon materials with different mass ratio of silica/chitosan(a,b), different pH value(c,d) and different reaction temperatures(e,f)
由图1(d)和表1可见,pH值从4.0增加到5.0,所制备介孔碳材料的平均孔径和介孔体积逐渐增大。这是因为,随着pH值从4增加到5时,TEOS的缩聚速率上升而水解速率下降[14],这可以提高胶体粒子的生长速率,使胶体颗粒的粒径逐渐增大。经碳化和硅模板去除后,碳材料的孔径和孔容逐渐增大。
由图1(f)和表1可见,当反应温度为20 ℃时,碳材料LPMC-1.33-4.5-20没有介孔,但含有少量微孔;随着温度增加到30 ℃和40 ℃时,所制备碳材料为典型的介孔材料,且在反应温度为30 ℃得到介孔碳LPMC-1.33-4.5-30的介孔孔容(2.56 cm3/g)最大。这是因为,Hollema[15]指出当反应温度为25 ℃时,烷氧硅烷与纤维二糖羟基的反应自由能接近0 kcal/mol。这意味着,较低的反应温度(20 ℃) 不能达到生成C-O-Si共价键的活化能,因此无法生成稳定的介观结构,得不到介孔。然而,当反应温度过高时,溶胶体系的稳定性降低[16],大尺寸的硅胶粒聚沉,导致40 ℃下制备的碳材料平均孔径和介孔体积较低。
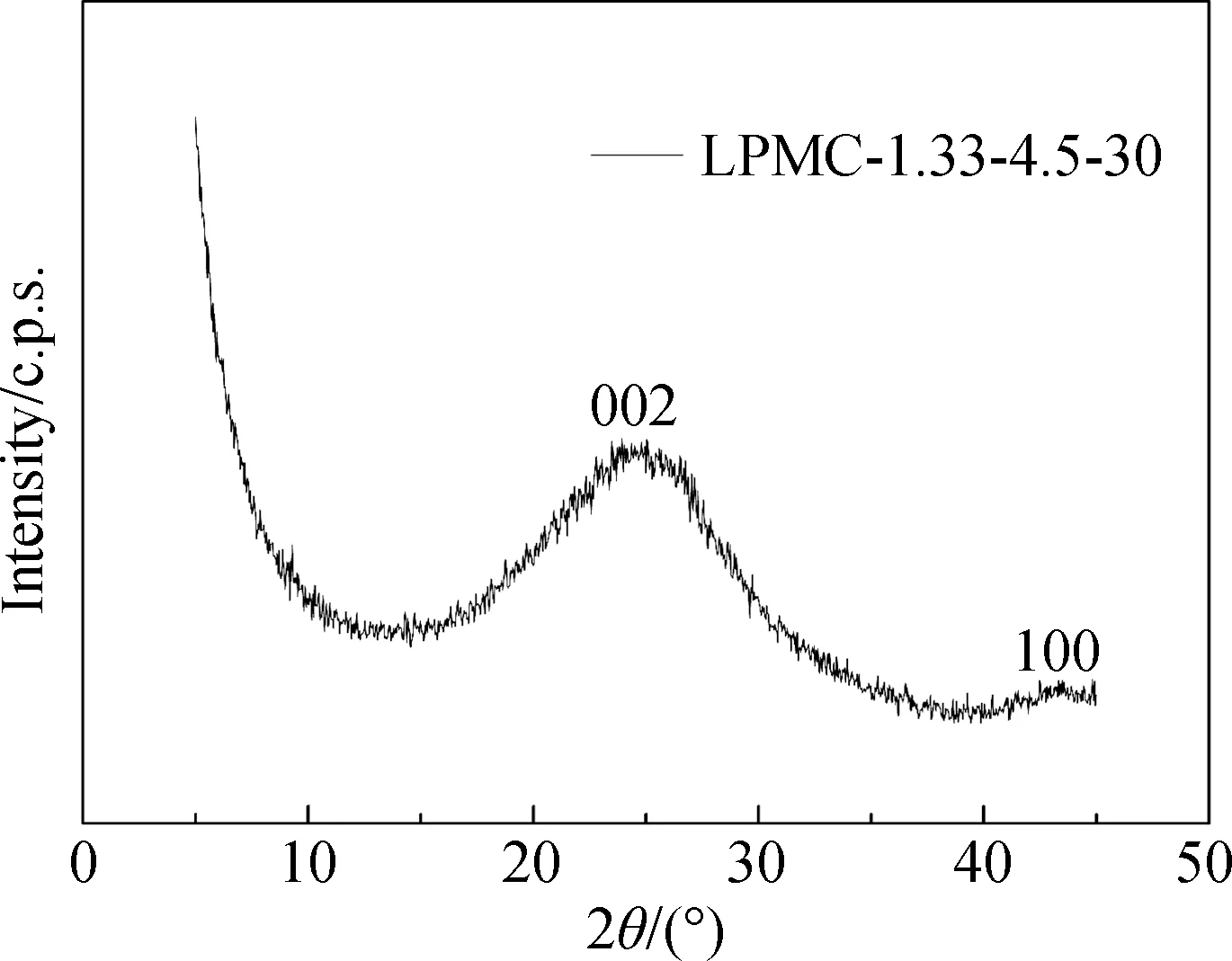
图2 LPMC-1.33-4.5-30的XRD图谱Fig.2 XRD pattern of the LPMC-1.33-4.5-30

图3 LPMC-1.33-4.5-30的SEM照片Fig.3 SEM image of the LPMC-1.33-4.5-30
3.2 大孔径介孔碳LPMC-1.33-4.5-30的形貌及组成分析
由于碳材料LPMC-1.33-4.5-30的介孔孔容和孔径最大,对其进行了XRD和SEM分析,数据分别如图2和图3所示。由图2的XRD图谱可见,碳材料在2θ为23°和43°出现两个较宽的衍射峰,为碳材料无定形结构的特征[17]。图3的SEM照片进一步说明碳材料表面的孔结构为典型的蠕虫状结构。
图4是碳材料LPMC-1.33-4.5-30的XPS光谱。由图4(a)可知,在样品表面检测到O、N、C元素,表明N元素成功原位掺杂到介孔碳材料中。图4(b)可见,该样品的N1s峰可拟合为四个峰,相应的能量分别集中在398.5 eV、399.9 eV、401.4 eV和402.8 eV,对应形态为N-6(吡啶氮)、N-5(吡咯氮)、N-Q(季铵氮)和N-X(与氧结合的吡啶氮)。

图4 LPMC-1.33-4.5-30的XPS谱图(a)和N1s XPS谱图(b)Fig.4 XPS(a) spectrum and N1s XPS spectra(b) of LPMC-1.33-4.5-30
表2为碳材料LPMC-1.33-4.5-30的元素分析。由表可见,碳材料表面主要存在C、N、O三种元素。利用XPS测得的碳材料上氮含量为3.95wt%。

表2 XPS测量的LPMC-1.33-4.5-30的元素组成Table 2 Elemental composition of LPMC-1.33-4.5-30 determined by XPS /wt%
3.3 大孔径介孔碳形成过程分析

图5 chitosan-silica-F127的Si2p XPS谱图Fig.5 Si2p XPS spectra of chitosan-silica-F127
图5是样品(未焙烧)的Si2p谱图。由图可见,Si2p峰经解叠加后得到结合能为102.5 eV和103.4 eV两个峰,分别对应于C-O-Si和Si-O-Si,前者是由于TEOS上的硅羟基和壳聚糖通过脱水反应生成新的C-O-Si共价键,后者是由于TEOS自身水解缩聚反应生成的Si-O-Si。
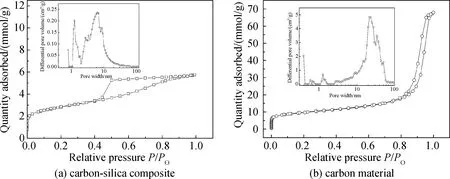
图6 制备的碳-硅复合物(a)和碳材料(b)的N2吸附/脱附等温线和孔径分布图Fig.6 N2 adsorption/desorption isotherms and pore size distributions of the obtained carbon-silica composite(a) and carbon material(b)
图6为焙烧后得到的碳-硅复合物(a)和对碳-硅复合物进一步除硅后得到碳材料(b)的N2吸附/脱附等温线及其孔径分布图。由图6(a)可见,N2的吸附容量很低,在P/P0>0.4时出现相对较小的滞后环,表明样品中存在少量介孔。根据DFT孔径分布,样品的孔径主要集中在7 nm,为高温碳化去除F127模板所致。由图6(b)可见,P/P0>0.8时,N2吸附量显著增加;由孔径分布可知孔径主要集中在20 nm附近,为进一步除硅模板所致。值得注意的是,本文制备的碳材料的孔径远远大于文献中的报道[18-19],这是由于论文采用HCl作为催化剂,H2O/TEOS摩尔比较大(75-110),TEOS水解缩聚行为与氨水催化类似[20],缩聚生成的硅氧链为三维网络状胶态聚合物,进一步缩聚形成粒径较大的胶粒;同时,由于碳源壳聚糖分子量较大,无法进入硅模板内部,其在共模板外面以C-O-Si共价键与TEOS结合(见XPS分析)。因此,碳化并除去硅模板后,得到大孔径和高孔容的碳材料。

图7 碳-硅复合物(a)和碳材料(b)的TEM照片Fig.7 TEM images of carbon-silicon composite(a) and carbon material(b)
图7是碳-硅复合物和碳材料的TEM照片。由图7(a)可见,碳-硅复合物中存在小于2 nm的微孔和2~7 nm的介孔,这是由于F127的去除所致,并注意到这些小孔是由硅模板包裹的。当进一步去除碳硅复合材料的硅模板时,可获得孔径较大的孔径为20 nm的碳材料(图6(b))。
因此,可以推断碳材料的形成过程:首先,形成以F127胶束为核、TEOS水解缩聚得到的硅胶粒为壳层的TEOS-F127共模板;然后,壳聚糖可以通过C-O-Si键与TEOS连接,包裹在共模板外。去除F127和TEOS后,获得了孔结构发达、孔径较大的介孔碳材料。
3.4 对BSA的吸附性能评价
图8和图9分别是碳材料LPMC-1.33-4.5-30对BSA分子的吸附速率曲线和吸附等温线。由图8可见,吸附时间低于4 h时,LPMC-1.33-4.5-30对BSA的吸附量急速增加,吸附量达到平衡吸附量的80.5%,这是由于初始阶段,样品的活性位点处于自由的状态,且较高的浓度差极大地促进了BSA的单分子层吸附。在4~12 h时,吸附量增幅减缓,这是由于吸附位点逐渐趋于饱和,出现BSA的多分子层吸附;在t=24 h时吸附基本达到平衡。可见,碳材料LPMC-1.33-4.5-30对BSA分子具有较高的吸附效率。
由图9可见,当BSA平衡浓度小于300 mg/L时,碳材料LPMC-1.33-4.5-30的平衡吸附量上升较快,主要是BSA分子在尺寸较均一的孔道内发生单分子层吸附;当BSA平衡浓度在300~500 mg/L范围内,平衡吸附量缓慢上升,主要是BSA分子在较大尺寸孔道内的堆积作用而发生多分子层吸附;当BSA平衡浓度大于500 mg/L后,平衡吸附量趋近平缓,吸附趋于平衡。
图10是运用Langmuir模型对吸附数据进行拟合得到的langmuir等温线,吸附方程如下:
Ce/qe=Ce/qm+1/(K·qm)
(2)
式中,qe为BSA在碳材料样品上的平衡吸附量,qm为碳材料对BSA的饱和吸附量,Ce为吸附平衡时BSA的浓度,K为BSA在37 ℃下的Langmuir吸附常数。结果显示,样品LPMC-1.33-4.5-30对BSA的吸附存在良好的线性关系,表明BSA分子在碳材料孔道的吸附均基本符合Langmuir单分子层吸附模型。通过计算得到LPMC-1.33-4.5-30样品对BSA饱和吸附量qm为344.8 mg/g。文献[21]测试了商用活性炭对BSA的吸附能力,结果表明,其吸附量仅为36 mg/g,远远低于论文制备的碳材料对BSA分子的吸附量。

图8 LPMC-1.33-4.5-30样品对BSA的吸附速率曲线Fig.8 The adsorption rate profiles of BSA molecule on the sample LPMC-1.33-4.5-30

图9 LPMC-1.33-4.5-30样品对BSA的吸附等温线Fig.9 The adsorption isotherms of BSA molecule on the sample LPMC-1.33-4.5-30
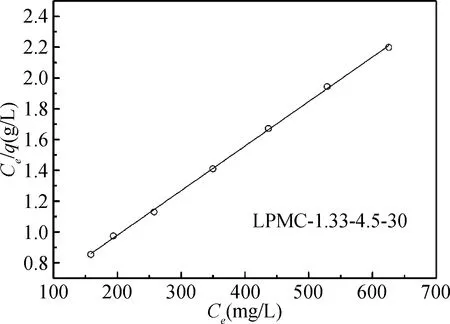
图10 LPMC-1.33-4.5-30样品对BSA的langmuir等温线Fig.10 Langmuir isotherm of BSA molecule on the sample LPMC-1.33-4.5-30
4 结 论
以壳聚糖为碳源、F127和TEOS为共模板成功一步合成了原位氮掺杂大孔径介孔碳。Si/C比、pH值和反应温度对碳材料的介孔体积和孔径分布都有重要影响。制备的介孔碳的平均孔径和介孔体积随Si/C和反应温度的升高先增大后减小,随pH值的增大而增大。当C/Si比为1.33、pH为4.5、反应温度为30℃时制备碳材料LPMC-133-4.5-30具有大的平均孔径(18.6 nm)和高的介孔孔容(2.8 cm3/g)。此外,以壳聚糖为碳源,氮元素成功原位掺杂到大孔径介孔碳材料中,其含量是3.95%,氮元素的掺杂形式主要为吡啶氮和吡咯氮等。
碳材料LPMC-133-4.5-30对BSA分子的吸附过程符合Langmuir单分子层吸附模型,饱和吸附量达到344.8 mg/g、4 h吸附量达到平衡吸附量的80.5%。表明碳材料对BSA分子具有较高的吸附效率。
