三周期性优选下的蛋白质表达区域预测算法研究∗
2019-10-08夏盛瑜辛月振陆志卿
夏盛瑜 辛月振 陆志卿
(中国石油大学(华东)计算机与通信工程学院 青岛 266000)
1 引言
DNA序列中带有遗传效应的片段称为基因,是遗传的结构和功能单位。真核生物的DNA序列中,如果把基因划分成许多分开的片段,那么把能够编码蛋白质的序列即基因的编码区,称作外显子;把不能编码蛋白质的基因序列称为内含子。序列要经过剪接、转录、复制过程才能合成蛋白质。而在合成蛋白质的过程中,外显子携带的遗传信息却得保存,准确地将遗传信息转递到蛋白质中,并参与指示各项生命活动。对编码区和非编码区的预测是基因预测的关键[1]。
弄清整个DNA序列中每一段基因所携带的遗传讯息对后续研究工作的进行起着关键作用,同时这也是人类研究的起初目的所在。由于真核生物的外显子和内含子之间缺乏明显的特征,基因组的蛋白质编码区域通常不连续[2]。
而原核生物,其蛋白质编码区域相当于真核生物的外显子区域。原核生物的基因预测已经有较多方法。总体可分为:1)基于同源性搜索的外部算法,称为间接遗传识别方法。现在最流行的序列对齐工具是FASTA和BLAST[3]。2)基于内在统计预测的算法。这种搜寻基因的方法与模式识别非常相似,分为基于隐式马尔科夫模型的经典的原核识别方法和基于神经网络的算法和基于曲线辨识分析的算法。3)混合法。它是内在算法和外部算法的组合。
根据3碱基周期理论,大多数外显子序列具有3碱基周期性,而内含子序列没有这个独特的特征。传统的三期分析方法将基因序列映射成数字序列,而蛋白质编码区域往往显示出较为明显的峰值。3周期性分析结合先验生物学知识,对预测结果有很好的解释。此外,功率谱分析方法具有良好的性能和良好的稳定性,因为需要比其他方法更少的参数,可以满足个人需要[5]。王其强利用三周期性对抑癌基因方面做了建模和算法分析[9]。最近,有学者利用韦尔奇函数法进一步完善了现有的功率谱分析方法,并提出了一种更有效的三次循环判断模型,并利用K-means聚类[17]和相关分析与3碱基周期结。因此信号处理方法也可用于基因组DNA研究,因为它们可以识别常规统计学方法无法检测的隐藏特征。将DNA序列转换为数字信号后,可以应用一些信号处理策略。作为生物蛋白序列的典型特征,V.R.Chechetkin于1995年已经对傅里叶谐波的大小和3碱基周期性的依赖关系做了讨论[4]。
在文中提出了一种可扩展的3碱基周期性预测方法来预测基因组蛋白编码区的优化方法。通过傅立叶分析,DNA序列转化为信号量。根据NCBI库中的实验验证蛋白质数,利用滑动窗口预测初始滑动框大小和阈值。对结果进行比较和筛选以缩小预测区域,然后使用可扩展滑动窗口策略来提高预测精度。
2 相关知识
2.1 三周期性
基因在编码蛋白质时是通过64个三联码(其中包含3个终止子)来确定20种氨基酸,蛋白质编码区具有周期3行为正是基于三联码的缘故[15]。即使许多外显子不具有周期三行为,但将它们连在一起编码蛋白质时大都表现出周期三行为。三周期行为可以表述为:将基因序列映射成数字序列,蛋白质编码区域往往显示出较为明显的峰值,这个峰值往往出现在序列的1/3和2/3处,而非编码区和随机序列则表现为无规则序列[16]。
由于直接处理DNA序列很困难,需要将DNA序列转化为数字信号,通过数值方法更方便处理DNA序列。这里使用的是Voss方法[7]。令S为基于长度为N的符号集I的DNA序列,其中I={A,T,C,G}。 DNA 序列 S可以由 S:S[0],S[1],…,S[N-1]表示,其中S[i]∈I。对于任何确定b∈I。
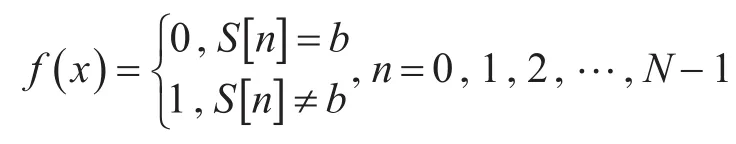
二进制序列uA(n),uT(n),uC(n),uG(n)的A,T,C和G的不论存在或不存在都可以在序列的第n位表示出来。
离散傅立叶变换(DFT)中,可以将信号序列转换为频域中的一组新值[6]。对于长度为N的信号序列,其DFT定义如下。
在k处的信号的DFT功率谱被定义为

DNA序列的DFT功率谱是其四个二进制指示符序列的功率谱之和。
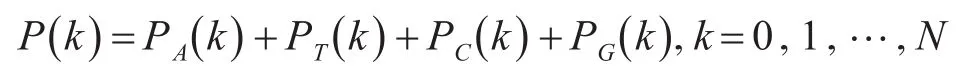
其中 PA(k),PT(k),PC(k)和 PG(k)是四个指示符序列 uA(n),uT(n),uC(n),uG(n)的傅里叶功率谱[12]。
2.2 滑动窗口
滑动窗口是基于DFT的一种优化,为了便于区分和识别功率谱。对于DNA序列S,其指示序列为{ub[n]}。长度M设置(设置为3的整数倍)为固定窗口的长度。对于以n(对于任何n(0≤n≤N-1)为中心的长度为M(n-(M-1)/2,n+(M-1)/2)的序列片段,当n接近开始和结束的DNA序列时,窗口实际有效长度可能小于M,那么可以有四个指令序列的 DFT[10]。

那么在M/3处的频谱值为p(n,M/3)。
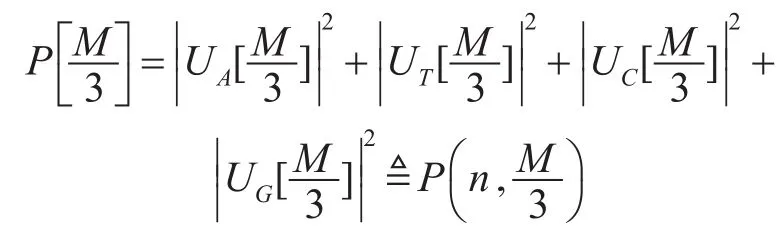
最后,使用归一化处理光谱值的结果。将结果除以频谱的最大值[11]。
3 相基于三周期性的蛋白质区域预测
3.1 基本外显子区域获取
频谱图的基础为三周期全序列功率图。在此基础上的频谱图是简化处理的一种实验手段。
图1和图2分别为实验中的编码区和非编码区的3种周期性功率谱的总和曲线。

图1 编码区三周期图

图2 非编码区三周期图
通过滑动窗口的算法在频谱中将不断出现较为明显的波动值,对预测有很好的帮助,对获取外显子区域是算法的基础。对于通过滑动窗口计算的长度为N的DNA序列,使用阈值A(用于区分外显子和其他编码区域),阈值B(用于子区域的控制长度)和频谱序列K的子区域发现外显子区域。为了提高傅里叶变换的效率,本文使用中的快速傅里叶变换[8](FFT)。
外显子获取的基本过程如下:
1)使用滑动窗口M计算DNA序列,得到P(n)的光谱序列;
2)设置K=0;
3)从起始点开始,P(n)与阈值A比较:如果P(n)> A,光谱序列可能区域K+1;
4)否则:如果光谱序列的子区域的长度大于阈值B,输出核苷酸序列;
5)否则:重新计数K;
6)增加n,重复步骤3)到步骤4)。
选取过程中,对各别异常点需要做包容处理,因为生物实验中不可避免的实验误差以及生物基因突变和实验环境的影响,外显子区域的获取尽量在一定容忍误差内。
在步骤4)中进行优化:
若P(n)<A,不再立刻重置K,而是暂时保存K值,判断之后β个位点值,若连续出现P(n)<A情况则重置K,若在β个位点内,仅出现δ个不满足情况,则此β段也为满足序列,即当前满足序列长度为K+β。
3.2 可扩展的滑动窗口
由于基因长度的不确定性,滑动窗口的大小应该是不相同的,以防止预测信息的遗漏。对于一个DNA序列,应该使用不同的滑动窗口来获得光谱。通过计算NCBI中的已知序列获得滑动窗口的大小。不同滑动窗口下的序列将使用外显子预测来获得大致的外显子区域。小滑动窗口的结果可能包括在大滑动窗口的结果中,因此确定该区域中外显子的可能尺寸。下面提出了一种新的优化策略:
1)重叠优先
重叠率越高意味着外显子存在的可能性就越大。所以优先选择高度重叠的区域,然后重叠区域的顺序将被合并。使用该序列作为两个末端的碱基序列和插入片段序列。使用等距滑动窗口操作来缩小外显子区域。重叠区域有很多可能性,但在实际情况中,重叠率并不是很高。当有三个或更多的重叠以满足条件时,进行进一步处理。
2)滑动框优化方法
在获得高重叠区域后也就获得该区域的滑动框范围,根据滑动框的起始大小,选择固定的步长对此区域进行不同大小的滑动框运算[13]。在不同的滑动框下获得的区域会逐渐稳定,即当到达头尾端点值误差在某个阈值范围内时。在此,若滑动框在高重叠区域的滑动框长度取值为[A,B],步长选取x,则需要进行(B-A)/x次的的滑动框运算,当高重叠区域很大时,次数会大大增加,影响预测效果。对此采用二分法思想:每次步长取当前滑动框长度差值的1/4,1/2,3/4,根据不同步长下的头尾端点阈值变化情况选取,可减少滑动框的运算次数。
4 实验结果
桑黄实验原始数据由中国石油大学(华东)生物中心提供,测试数据为Fasta格式纯文本文件。这些文件中的序列大小为初选数据为5.4G,测序公司统计优选文本45M,通过本文方法获取的可能序列与NCBI库进行了比对,其中与Translation elongation factor 1 alpha和ATP synthase subunit a蛋白质有较好的匹配度,最高分为356,E值为3e-99,Ident为85%,表明算法有较好的可靠性。整体算法首先预处理文本数据;然后选择阈值(阈值包括滑动窗口的大小和外显子区分值);通过区域映射和滑动窗口操作DNA序列获取原始频谱图;接着对外显子区域进行预测;最后使用可扩展的滑动框策略缩小预测区域直至满足阈值条件如表1所示(单位是碱基个数)。

表1 阈值选择
滑动框大小的选取结合一些先验知识,并结合NCBI库中鞘氨醇单胞菌的公开数据中已知基因长度进行的数理统计结果。在NCBI库中,通过固定滑动窗口分析不同长度的参考序列,并在序列的两端添加背景序列的固定长度。计算序列的头部和尾部的光谱值。光谱如图3所示。对头尾光谱的计算统计获得符合实验要求的阈值。
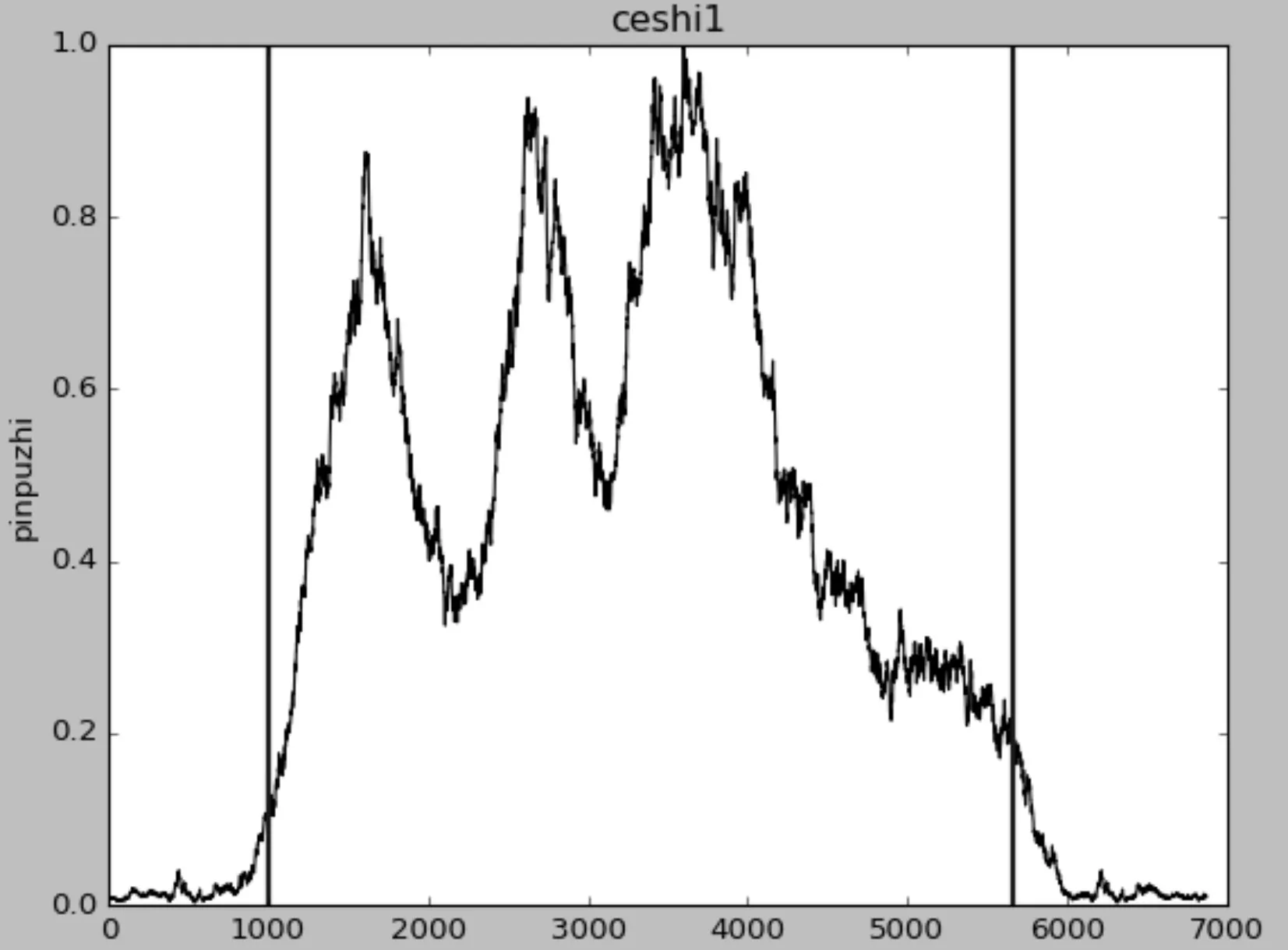
图3 频谱图中的光谱值
由于NCBI和Uniport等公开数据库中的桑黄基因较短,所以测试桑黄二级产物的Phosphoenolpyruvate carboxylase蛋白质ppc基因,888个氨基酸序列,预测蛋白质编码区的阈值为0.2。在大多数情况下测试结果表现良好,特别是当序列较长时。在本文中,不使用传统的经验阈值0.4,因为不同的生物具有不同的遗传结构特征。不同的窗口尺寸,不同的功率谱密度计算方法也将导致功率谱预测曲线幅度的变化。因此,经验阈值将带来显着的预测偏差(在下图中背景序列下的起始位点为1000)。

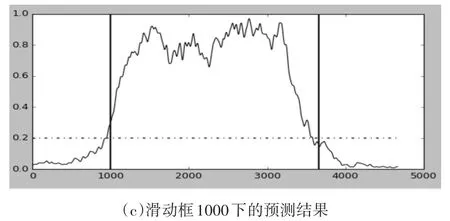
图4
为了测试提出的方法,本文添加了仿真实验。从Uniprot数据库中随机选择5种桑黄及其近缘的蛋白质,并将它们经过指定次数的变异后插入到长的背景序列中。经过粗略的外显子预测后,不同滑动窗口的可能区域的数量如下表所示。然后使用端点对齐和合并重叠序列,并选择了两个新的序列,对于新序列1,使用300到100个碱基长度的等距滑动窗口来缩小区域并删除了非表达区域。对于新序列2,使用等距滑动窗从500到200个碱基长度。根据头尾位点反馈信息,选取新的滑动框。

表2 背景序列下的测试结果
最后得到两个可能的区域,预测结果与其中一个插入序列有良好的匹配度。而不经过优化的算法获得的初步区域数量较多,虽然包含了目标序列,但是缺乏实际应用的意义。特别是滑动框为100和200的序列,其数量较多,但是只是滑动框为500的初选结果的一部分,重叠区域过多,影响精确度。
5 结语
本文针对生物的蛋白表达区域问题,提出了一种基于3碱基周期性蛋白表达区域的优化方法。实验中,功率谱分析方法的算法性能良好,方法所需的各种参数设置比其它方法少,稳定性好[14]。应用通过已知的DNA序列和实际实验数据的预测结果经过Blast后,部分结果数据和NCBI中的已知数据相符,有些将具有进一步验证的生物实验。通过这些结果比对,验证此方法能良好地预测蛋白质编码区。其中可扩展的滑动窗口是一个重要的部分,使用区域选择优化策略降低数据复杂度,缩小可能区域。在下一步的研究中,将通过添加自适应滑动窗口来减少人为的主观干预,使该方法的预测精度进一步地提高。时间复杂度和误差检验[18]也是未来优化的方向。
