液相色谱串联质谱法测定不同茶叶中的氯噻啉
2019-09-24徐潇颖罗金文刘柱金绍强朱炳祺
徐潇颖 罗金文 刘柱 金绍强 朱炳祺
摘要[目的]建立茶叶中氯噻啉残留量的液相色谱-串联质谱检测方法。[方法]茶叶样品经乙腈提取,提取液通过TPT固相萃取柱净化后进行仪器分析。对样品提取、净化及仪器条件参数进行优化。[结果]氯噻啉在1 ~500 ng/mL线性关系良好(R2 >0.999);检出限为1.5 μg/kg,定量限为5.0 μg/kg;在0.005、0.010、0.050和3.000 mg/kg条件下,回收率可达88.8%~109.1%,相对标准偏差(RSD)≤54%(n=6)。[结论]通过方法学考察和实验室间验证表明,该方法灵敏、准确,适用于各种茶叶中氯噻啉残留量的定性和定量分析。
关键词 氯噻啉;液相色谱-串联质谱;茶叶;固相萃取
中图分类号 TS207.5+3文献标识码 A
文章編号 0517-6611(2019)15-0201-04
doi:10.3969/j.issn.0517-6611.2019.15.055
开放科学(资源服务)标识码(OSID):
Abstract[Objective]The research aimed to establish a liquid chromatographytandem mass spectrometry method for the determination of chlorothiazide residues in tea.[Method]Tea sample was extracted by acetonitrile and purified by TPT solid phase extraction before instrumental analysis. This study optimized extraction method, clean up method and instrumental parameters.[Result]The linear relationship of chlorothiazide in 1-500 ng/mL is good (R2 > 0.999);the limit of detection (LOD) and limit of quantification (LOQ) were 1.5 μg/kg and 5.0 μg/kg, respectively. The recoveries were 88.8%-109.1% with relative standard deviations (RSD) lower than 5.4%(n=6).[Conclusion]Based on the results of method validation and interlaboratory validation, this method has proven to be sensitive, accurate and suitable for the qualitative and quantitative determination of imidaclothiz residue in tea.
Key words Imidaclothiz;Liquid chromatographytandem mass spectrometry(LCMS/MS);Tea;Solid phase extraction
基金项目 国家重点研发计划项目(2018YFC1603400)。
作者简介 徐潇颖(1989—),女,浙江金华人,工程师,硕士,从事食品质量安全研究。*通信作者,工程师,博士,从事食品质量安全控制研究。
收稿日期 2019-03-05
氯噻啉是我国自主研发的一种新型烟碱类杀虫剂,它通过作用于烟酸乙酰胆碱酯酶受体以阻滞害虫的中枢神经正常传导,其不仅可用于防治吮吸式口器害虫,同时对鞘翅目、双翅目和鳞翅目害虫也有效,尤其对水稻二化螟、三化螟毒力比其他烟碱类杀虫剂高,可以广泛用于水稻、小麦、蔬菜、烟草、棉花、果树、茶树等作物,具有高效、光谱、低毒等优点[1-2]。由于氯噻啉对哺乳动物毒性较低,被国家工信部列为“十一五”高度农药转产和替代项目,具有一定的应用前景[3]。
我国最新修订的GB 2763—2016《食品安全国家标准食品中农药最大残留限量》中规定了茶叶中氯噻啉的最大残留量为3.0 mg/kg,但国内尚未制定关于茶叶中氯噻啉农药残留量检测的标准方法。目前已报道的关于氯噻啉的检测方法主要有液相色谱法[4]、酶联免疫法[5]、液相色谱-串联质谱法[6]和气相色谱-质谱法[7]。施海燕等[8]利用生物素-链霉亲和素的高亲和作用,研制了一种简便、快速的氯噻啉胶体金增强免疫层析分析方法,但该方法灵敏度差,容易出现假阳性。贺敏等[4]用二氯甲烷提取水稻样品中的氯噻啉,提取液浓缩后通过佛罗里硅土固相萃取柱进行净化,使用液相色谱法检测。杨东冬等[9]制备分子印迹固相萃取,使用液相色谱法测定大米中的氯噻啉,回收率在81.2%~97.5%。刘松南等[6]采用QuEChERS前处理方法,使用液相色谱-串联质谱法测定茶叶中氯噻啉。相较于其他检测方法,液相色谱-串联质谱法具有高选择性、高灵敏度的特点,能有效检测目标化合物。但是液相色谱-串联质谱法在定量分析过程中容易受到基质的影响,不仅污染仪器,而且影响分析检测的准确性。在日常检测过程中,茶叶基质复杂,较谷物类食品和水果类食品含有更多的色素等基质干扰物,因此需要有效的前处理方法以保证检验结果的准确性。
针对在农业生产过程中氯噻啉使用的推广和有效检测方法缺失的矛盾,该研究对茶叶中氯噻啉的前处理方法及仪器测定条件进行优化,并对建立的方法进行方法学考察和实验室间验证,建立准确可靠的检测方法。
1 材料与方法
1.1 仪器
LC-MS 8050液相色谱-串联质谱仪:配有电喷雾离子源(ESI)(日本Shimadzu公司)。Mettler XPE 205分析天平(瑞士Mettler-Toledo公司)。Multi-Prep自动均质器(美国PRO Scientific公司)。离心机(美国Thermo Fisher公司)。旋转蒸发仪(德国IKA公司)。
1.2 试材
乙腈(质谱纯)、二氯甲烷(色谱纯)购自德国Merck公司;甲苯(色谱纯)购自美国TEDIA公司;甲酸(质谱纯)购自美国Fisher Scientific公司。氯化钠(分析纯)、无水硫酸钠(分析纯)购自国药集团化学试剂有限公司。氯噻啉标准品购自农业部环境保护科研监测所,浓度为100 μg/mL,CAS号为105843-36-5;Cleanert TPT固相萃取柱(1 g,6 mL)、N-丙基乙二胺(PSA)由天津博纳艾杰尔公司提供;试验用水均为经过Milli-Q系统净化后的去离子水。
1.3 试验条件
1.3.1 标准溶液的配制。
氯噻啉标准品用乙腈稀释成浓度为5 μg/mL的标准品中间液。准确移取中间液,用经过前处理的空白基质溶液稀释配制成浓度为1、20、50、100、200、500 ng/mL的基质标准曲线工作液。
1.3.2 液相色谱-串联质谱方法。
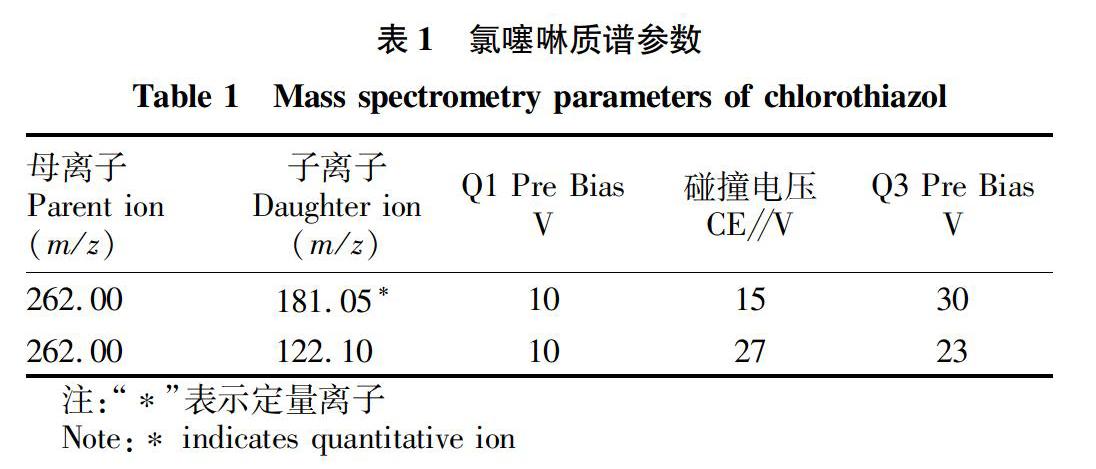
离子源:电喷雾离子源(ESI)。扫描方式:正离子扫描。毛细管电压4 kV。毛细管温度300 ℃。雾化气:氮气,3 L/min。碰撞气:氩气。检测方式:多反应监测(MRM)。质谱参数见表1。色谱柱:Agilent Eclipse Plus C18(2.1 mm×50 mm,1.8 μm);柱温30 ℃;进样量1 mL;流动相A:0.1%甲酸水,B:乙腈;流速0.25 mL/min;梯度洗脱程序:0~3.0 min,15% B;3.0~6.0 min,15% B~75% B;6.0~8.0 min 75% B;8.0~8.1 min,75% B~15% B;81~10.0 min,15% B。
1.3.3 样品处理。
1.3.3.1 提取。
称取2 g粉碎均匀样品(精确至0.001 g),置于50 mL离心管中,加入15 mL乙腈,涡旋振荡0.5 min,在高速组织捣碎机上以15 000 r/min匀质1 min后4 000 r/min离心5 min,将上清液移入梨形烧瓶中。残渣再用15 mL乙腈,重复上述提取过程一次,合并所有上清液,在40 ℃水浴中减压旋转浓缩至2~3 mL。
1.3.3.2 净化。
在TPT固相萃取柱中加入约2 cm高无水硫酸钠并预先用5 mL乙腈-二氯甲烷(体积比3∶1)活化固相萃取柱。将浓缩提取液转移至固相萃取柱上,再用20 mL乙腈-二氯甲烷(体积比3∶1)洗脱,收集洗脫液于梨形烧瓶中,在40 ℃水浴中减压旋转浓缩至近干,氮吹至干。用2.0 mL乙腈溶解残渣,经0.22 μm滤膜过滤后,供液相色谱-质谱测定。对于含量较高的样品,用空白基质溶液稀释乙腈复溶液后,供仪器测定。
2 结果与分析
2.1 仪器条件优化
在液相色谱串联质谱仪上氯噻啉的检测模式为正离子模式,在方法开发过程中,依次考察了甲酸铵、乙酸铵和是否含有甲酸等分别作为水相时氯噻啉的响应强弱,结果显示,氯噻啉的响应由强到弱依次为水>0.1%甲酸水溶液>5 mmol/L甲酸铵水溶液>含0.1%甲酸的5 mmol/L甲酸铵水溶液>含0.1%甲酸的5 mmol/L乙酸铵水溶液>5 mmol/L乙酸铵水溶液。尽管添加甲酸会抑制化合物的响应,但是试验结果显示纯水做流动相时容易产生峰前移并影响峰形。此外在乙腈中添加0.1%的甲酸对目标物的响应值未有显著提高。因此最终选择0.1%甲酸水-乙腈作为此次分析的流动相体系,采用梯度洗脱的方法可以将残留在色谱柱上化合物充分洗脱。
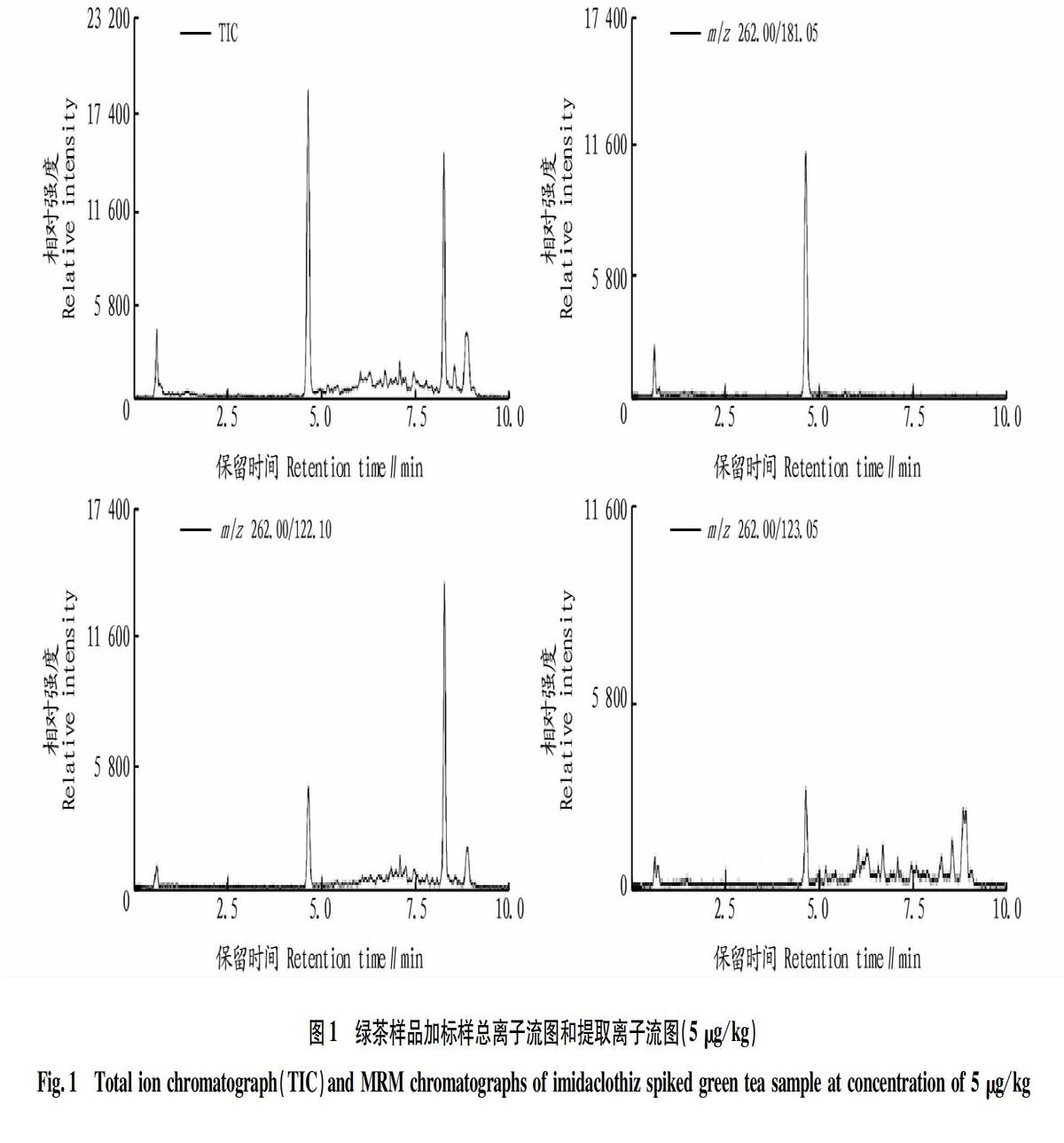
在色谱柱的选择中,分别考察了安捷伦Eclipse Plus C18(2.1 mm×50 mm,1.8 μm)、安捷伦Poroshell 120 EC-C18(3.0 mm×100 mm,2.7 μm)、安捷伦ZORBAX Eclipse XDB-C18(2.1 mm×50 mm,1.8 μm)、Thermo Hypersil GOLD(2.1 mm×100 mm,3 μm)4种不同型号和规格的反相色谱柱,采用上述选定的流动相条件进行分离,结果表明绝大多数的C18色谱柱对氯噻啉均有保留,根据色谱柱长短适当调整流动相条件均可满足试验要求。该研究选择安捷伦Eclipse Plus C18(2.1 mm×50 mm,1.8 μm),色谱图见图1。
试验过程中采用自动优化功能对质谱系统各参数进行快速优化,确定特征母离子和子离子。试验得到母离子为m/z 262.0,二级碎片离子主要有m/z 181.05、122.10、123.05等。进一步优化二级碎片离子的碰撞电压等参数,根据响应强度和特异性,选取 m/z 262.00/181.05为定量离子,m/z 262.00/122.10为定性离子,优化得到碰撞电压分别为15和27 V。
2.2 提取条件的选择
氯噻啉易溶于大部分有机溶剂,乙腈、二氯甲烷、甲醇、丙酮均为良好的提取溶剂,试验结果显示采用上述溶剂进行提取后,所得回收率均能达到80%以上。相比于丙酮、二氯甲烷等,乙腈作为提取溶剂时,保证较好的提取效果的同时可以减少基质干扰物的溶出[10]。此外在提取过程中,部分方法会采用加水对茶叶进行浸泡后再进行乙腈提取。如图2所示,经过水浸泡后的茶叶样品提取液在净化后颜色明显深于未经水浸泡的茶叶样品。水的浸泡过程容易将茶叶的水溶性色素提取出来,从而带来更多基质干扰[11]。因此,在此方法中选择乙腈直接提取茶叶样品。
在提取方式上,通常有超声、涡旋、振摇和均质的方式,均质可将样品基质中细胞等组织结构破坏并充分地混合均匀[12],为了保证能有效地将样品中的待测目标物提取到溶剂中,故采用均质方法进行提取。
2.3 净化条件的优化
在检测过程中,QuEChERS方法[13]有着操作便捷、适用范围广的特点,但多用于蔬菜水果的净化。当基质变成茶叶、韭菜等样品时,PSA吸附剂对色素净化的效果有限,TPT固相萃取内填复合吸附材料,在去除色素等基质干扰物上有更好的效果。在研究中发现TPT固相萃取柱对氯噻啉有一定的吸附作用,使用乙腈不能有效地将目标物洗脱下来,导致回收率低于80%。因此净化过程中采用乙腈-二氯甲烷(体积比3∶1)作为洗脱液以有效洗脱目标物,此外试验对比了乙腈-甲苯(体积比3∶1)混合溶液作为洗脱溶液时的洗脱效果。尽管试验结果显示使用2种洗脱液回收率均达到90%以上,但如图2所示,使用乙腈-甲苯(体积比3∶1)作为洗脱液时得到的样品溶液颜色深于使用乙腈-二氯甲烷(体积比3∶1)作为洗脱液时得到的样品。另一份试验采用QuEChERS净化方法,参考AOAC 2007.01,茶叶样品定量加入10 mL乙腈提取液,在每毫升乙腈提取液中加入100 mg的PSA粉末进行净化。图2试验结果显示经过该QuEChERS方法得到的样品溶液颜色明显深于使用TPT固相萃取柱方法得到的样品溶液。因此,在该方法中选择使用TPT固相萃取柱净化方法并使用乙腈-二氯甲烷(体积比3∶1)作为洗脱溶剂,可以较好地去除色素杂质。
47卷15期 徐潇颖等 液相色谱串联质谱法测定不同茶叶中的氯噻啉
2.4 方法学验证
2.4.1 标准曲线的建立。
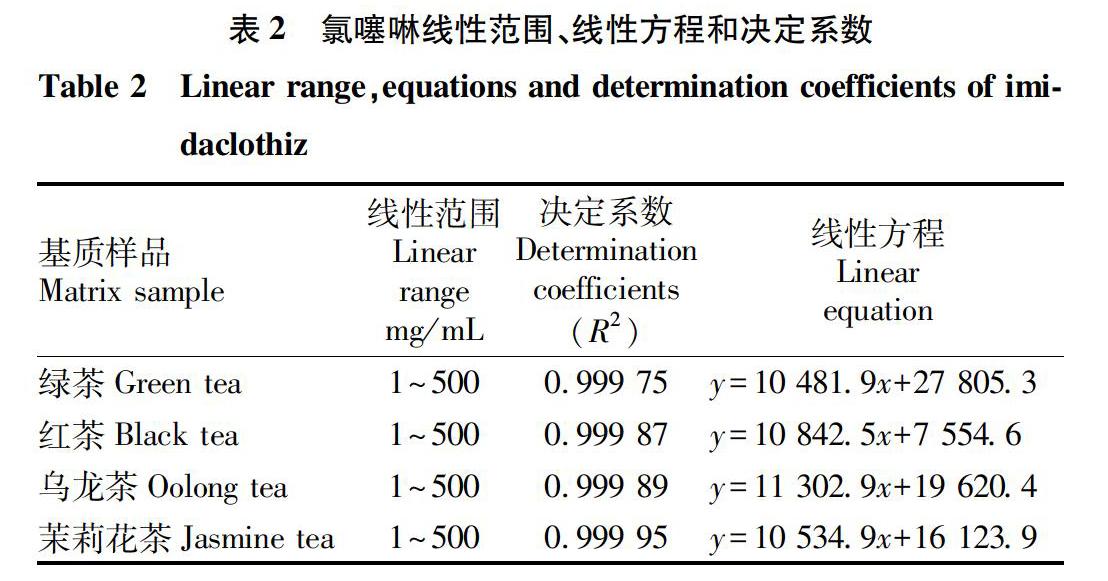
依据“1.3.1”配制基质标准曲线工作液并上机测试,以响应值(Y)与目标物浓度(X)建立线性拟合方程。经验证,在1~500 ng/mL以不同基质配制的标准曲线线性关系良好,决定系数R2不小于0.999(表2)。
2.4.2 精密度、回收率及检出限。
在阴性茶叶样品中加入低、中、高、限量值4个不同浓度的标准工作溶液进行加标回收与精密度测试,结果见表3。数据表明,4个加标水平下的回收率为88.8%~109.1%,6次平行测试的相对标准偏差为1.5%~5.4%。
在空白茶叶样品中加标并进行检测,当定量离子和定性离子色谱峰的信噪比(S/N)≥3时,样品中对应的氯噻啉含量为1.5 μg/kg,为该方法的检出限。当定量离子色谱峰S/N≥10时,样品中氯噻啉的含量为 5.0 μg/kg,为该方法的定量限。
2.4.3 基质效应。
基质效应对质谱的离子化效果产生影响,从而对结果产生影响,因此为重要评价依据之一[14]。通常以基质匹配标准曲线和溶剂标准曲线的斜率比值,作为基质效应(ME),当ME>1.1时为基质增强效应,而ME<0.9时为基质抑制效应[15]。分别将红茶、绿茶、乌龙茶和茉莉花茶样品基质匹配标准曲线与乙腈配制的校正曲线进行比较,试验结果显示在4种茶叶基质中,基质效应均为0.88~0.92。为保证结果准确性,试验过程中采用基质校正曲线对样品进行校正。
2.5 实验室间方法验证
实验室之间不同仪器条件和不同实验人员的操作会对试验结果产生一定影响,为了保证方法的可重复性,选取红茶、绿茶、茉莉花茶样品在另外5家实验室进行方法验证,验证内容包括灵敏度、校正曲线、线性范围、回收率试验。结果显示,在1~500 ng/mL,不同基质标准曲线的线性关系良好,R2均不小于0.999。在1.5和5.0 μg/kg浓度下满足S/N≥3和S/N≥10。在 0.005、0010、0.050 mg/kg 3个加标浓度下加标回收率均在70%~120%,RSD≤7.9%。
2.6 实际样品测试
将该研究建立的方法,应用于市场上流通的预包装茶叶的检测,其中绿茶15批、红茶9批、乌龙茶6批、花茶4批,均未检出氯噻啉。
3 结论
该研究结合了TPT固相萃取的前处理方法和液相色谱串联质谱仪器分析方法,建立了茶叶中灵敏、准确的氯噻啉测定方法。试验结果显示茶叶样品直接使用乙腈提取,通过TPT固相萃取柱方法并使用乙腈-二氯甲烷(体积比3∶1)为洗脱液,能保证回收率的同时达到较好的净化效果。方法学验证结果和实验室间验证结果均满足GB/T 27404—2008 《实验室质量控制规范 食品理化检测》的相关规定,为不同类型的茶叶样品中氯噻啉残留量的定性和定量分析提供有效的检测手段。
参考文献
[1]贺敏,贾春虹,陈莉,等.高效液相色谱法测定大白菜中虫酰肼和辛硫磷的残留量[J].农药,2010,49(1):50-52.
[2]戴宝江.新颖杀虫剂——氯噻啉[J].世界农药,2005(6):46-47.
[3]张敏恒,赵平,严秋旭,等.环己烯酮类除草剂市场开发[J].农药,2012,51(12):859-862.
[4]賀敏,贾春虹,余平中,等.土壤和葵花籽中精异丙甲草胺的残留分析[J].农药,2009,48(4):285-286.
[5]方松,张斌,施海燕,等.氯噻啉酶联免疫分析方法研究[J].农药学学报,2011,13(3):287-292.
[6]刘松南,赵新颖,董晓倩,等.QuEChERS净化-液相色谱-串联质谱法测定茶叶中氯噻啉[J].色谱,2015,33(11):1205-1209.
[7]许秀莹,施海燕,王鸣华.气相色谱-质谱联用测定大米中6种烟碱类农药残留[J].质谱学报,2012,33(2):99-103.
[8]施海燕,盛恩泽,马明,等.气相色谱-质谱联用测定大米中6种烟碱类农药残留[J].分析化学,2017,45(3):403-408.
[9]楊东冬,丛路静,田明明,等.分子印迹固相萃取-液相色谱联用法测定3种新烟碱类农药的残留[J].分析化学,2014,42(6):872-877.
[10]余璐,宋伟,吕亚宁,等.超高效液相色谱-四极杆-飞行时间质谱法快速筛查茶叶中的204种农药残留[J].色谱,2015,33(6):597-612.
[11]张媛媛,张卓,陈忠正,等.QuEChERS方法在茶叶农药残留检测中的应用研究进展[J].食品安全质量检测学报,2014,5(9):2711-2716.
[12]陆小磊,叶美君,周卫龙.超高效液相色谱-串联质谱法测定茶叶中氯噻啉的残留量[J].农药,2014(11):825-828.
[13]ANASTASSIADES M,LEHOTAY S J,STAJNBAHER D,et al.Fast and easy multiresidue method employing acetonitrile extraction/partitioning and "dispersive solidphase extraction" for the determination of pesticide residues in produce[J].Journal of AOAC International,2003,86(2):412-431.
[14]ZHAO P Y,WANG L,ZHOU L,et al.Multiwalled carbon nanotubes as alternative reverseddispersive solid phase extraction materials in pesticide multiresidue analysis with QuEChERS method[J].Journal of chromatography A,2012,1225:17-25.
[15]LOZOWICKA B,ILYASOVA G,KACZYNSKI P,et al.Multiresidue methods for the determination of over four hundred pesticides in solid and liquid high sucrose content matrices by tandem mass spectrometry coupled with gas and liquid chromatograph[J].Talanta,2016,151:51-61.
