Application of GLAD-PCR Assay for Study on DNA Methylation in Regulatory Regions of Some Tumor-Suppressor Genes in Lung Cancer
2019-09-24SmetannikovaEvdokimovNetesovaAbdurashitovAkishevDubininPozdnyakovVihlyanovNikitinTopolnitskyKarpovKolomietsKhDegtyarev
N.A. Smetannikova, A.A. Evdokimov, N.A. Netesova, M.A. Abdurashitov, A.G. Akishev, E.V.Dubinin, P.I.Pozdnyakov, I.V. Vihlyanov, M.K. Nikitin, E.B. Topolnitsky, A.B. Karpov, S.A. Kolomiets, S.Kh. Degtyarev
1State Research Center of Virology and Biotechnology, Koltsovo, Russia; 2EpiGene LLC, Novosibirsk, Russia; 3Altai Regional Oncology Center, Barnaul, Russia; 4Tomsk Regional Clinical Hospital, Tomsk, Russia; 5Seversk Biophysical Research Centre,Seversk, Russia; 6Regional Clinical Oncology Center, Kemerovo, Russia
Abstract Hypermethylation of the gene regulatory regions are common for many cancer diseases. In this work we applied GLAD-PCR assay for identificating of the aberrantly methylated RCGY sites in the regulatory regions of some downregulated genes in tissue samples of lung cancer (LC). This list includes EFEMP1, EPHA5, HOXA5, HOXA9, LHX1, MYF6, NID2, OTX1, PAX9, RARB,RASSF1A, RXRG, SIX6, SKOR1 and TERT genes. The results of DNA samples from 40 cancer and 25 normal lung tissues showed a good diagnostic potential of selected RCGY sites in regulatory regions of MYF6, SIX6, RXRG, LHX1, RASSF1A and TERT genes with relatively high sensitivity (80.0 %) and specificity (88.0 %) of LC detection in tumor DNA.
Key words Lung neoplasms; DNA methylation; GLAD-PCR; Diagnostics; Epigenetics
Introduction
Lung cancer (LC) is one of the major malignancies annually leading to more than 1.5 million cases of cancer death worldwide[1,2]and requiring an early diagnostics method.
The detection of methylation of RCGY sites in tumorsuppressor gene regulatory regions, which occurs in the tumor cell DNA at the initial stages of the disease, is one of the most promising diagnostic and prognostic tools. Such an aberrant methylation is typical for the most of sporadic cancers at early stages, constituting more than 90% of all malignant neoplasms[3,4].
It is well known that de novo DNA methylation,including abnormal DNA methylation in cancer cells,is performed by DNA methyltransferases Dnmt3a and Dnmt3b, which predominantly recognize RCGY site(whereas R is A or G, Y is T or C) and modify it with formation of R(5mC)GY sequence in both DNA strands[5].Methyl-directed DNA endonuclease GlaI specifically recognizes and cleaves R(5mC)↓GY sites as indicated by the arrow[6]. Due to this unique specificity it is possible to use GlaI for identification of the tumor cell DNA.
Based on this enzyme we have previously developed a GLAD-PCR assay for detection of R(5mC)GY sequence in a definite position of human genome even at significant excess of DNA molecules with this non-methylated site(RCGY)[7]. The assay includes DNA hydrolysis with GlaI,ligation of the obtained DNA fragments with the specific adapter followed by real-time PCR with TaqMan probe and a genomic primer, which are complementary to target DNA fragment, and the second primer, which is complementary to the adapter sequence.
Recently, we used GLAD-PCR assay to analyze the tumor-suppressor genes methylation in DNA samples from colorectal cancer tissues[8,9]. In the present work we apply GLAD-PCR assay to detect an aberrant methylation of RCGY sites in the regulatory regions of tumor-suppressor genes in DNA samples from tumor tissues in case of LC.
Materials and methods
The study specimens were bronchopulmonary tumor DNA samples, obtained from 40 LC patients who had undergone surgery. The most of patients had squamous-cell lung carcinomas (n=19) or adenocarcinomas (n=13) with varying degrees of differentiation. In other cases small-cell carcinomas (n=2), large-cell carcinomas (n=2) and mixed carcinomas (n=2) (squamous-cell carcinoma combined with adenocarcinoma and large-cell carcinoma combined with neuroendocrine tumor) were diagnosed. Meanwhile five patients had clinical stage I (T1-2aN0M0), 12 had stage II (T1-3N0M0, T1-2N1M0), 18 had stage III (T1-2N2M0,T3N1-2M0, T4N0-2M0, T1-4N3M0) and four patients were diagnosed with stage IV (distant metastases (M1) in any primary tumor size (T) and metastatic status of the regional lymph nodes (N)). In one case the malignant neoplasm was the local relapse of squamous-cell lung carcinoma with low differentiation degree.
As a control, we used DNAs from normal lung tissues,obtained from 25 LC patients during the surgery at a considerable distance from the tumor focus, on the resection line.
The aim of this study was preliminary selection of R(5mC)GY sites - potential epigenetic markers of lung cancer which will be further verified on wider number of samples obtained from blood cell-free DNA.
All participating patients voluntarily joined this study with the written informed consent.
The preservation procedure and storage conditions for tissue samples are described previously[8]. DNA isolation and purification were performed using the standard phenolchloroform method[10].
DNA samples were analyzed by GLAD-PCR assay as described previously in detail[9].
To design specific primers and probes, we used the nucleotide sequences (human genome version GRCh38/hg38) from the GenBank database (http://ncbi.nlm.nih.gov/genbank), Vector NTI 11.5 software (Invitrogen, USA),and NCBI BLAST resource (http://blast.ncbi.nlm.nih.gov).A list of primers and probes is provided in Table 1. Hybrid primers, corresponding to the studied RCGY sites, were selected as described previously[8,9].
The statistical analysis of experimental data was performed using the MedCalc 15.11 software (MedCalc Software, Ostend, Belgium). According to the quantification cycle (Cq) values obtained for each studied RCGY site,receiver operating characteristic (ROC) curves with 95% confidence interval (CI) were determined to assess the assay sensitivity and specificity with an area under the ROC curve (AUC) being estimated nonparametrically. AUC is the cumulative indicator of the marker diagnostic efficacy (for the “perfect” test AUC = 1)[11].
Results
The determination of candidate tumor-marker genes in lung cancer. When searching for LC epigenetic markers,DNA samples from the tumor and normal lung tissues are mainly examined. The comparison of gene methylation status in tumor and normal cells enables to identify potential epigenetic tumor markers that are much more frequently methylated in malignant transformation.
As follows from the recent studies, a number of candidate epigenetic LC determinants have been identified using bisulfite DNA conversion method. We chose 15 tumorsuppressor genes in regulatory regions of which we studied methylation of RCGY sites using GLAD-PCR assay. This list includes MYF6, SIX6 и RARB PΛ[12], NID2 и RASSF1A[13],TERT и RASSF1A[14], EFEMP1[15], EPHA5[16], HOXA5[17],HOXA9[18], LHX6[19], OTX1[20], PAX9[21], RXRG[22]и SKOR1 genes[23].
Identification of RCGY sites in DNA from LC tissues suitable for GLAD-PCR analysis. Screening of the mostly methylated RCGY sites in regulatory regions of tumorsuppresor genes was carried out as described earlier[8,9]using 10 DNA samples. For each RCGY site in the studied DNA region (~200 nucleotides from the genomic primer hybridization site) we used corresponding hybrid primer.A criterion of the site selection was the lowest value of the threshold cycle (Cq) obtained in GLAD-PCR assay.
The selected R(5mC)GY sites and corresponding hybrid primers, which were used for further study of the samples DNA collection (both LC and normal tissue samples), are provided in Table 2.
GLAD-PCR assay of RCGY sites in DNAs from the clinical samples. GLAD-PCR assay of selected sites in DNAs, obtained from LC samples (n=40) and normal lung tissue (n=25) was performed in triplets. There was 3 ng of DNA (~103copies of the studied gene region) in the reaction mixture. Figure 1 presents the diagrams of the average values of threshold amplification cycles (Cq) obtained by GLADPCR analysis of RCGY sites located in LC tumor-suppressor genes.
The statistical analysis of the results of GLAD=PCR assay(Figure 2 and Table 3) allows to draw a conclusion about the good diagnostic characteristics of the RCGY sites in tumorsuppressor genes MYF6, SIX6, RXRG, LHX1, RASSF1A and TERT: obtained AUC values for these markers are in 0.727-0.805 range[11]).
For these six LC tumor-markers, the total diagnostic characteristics were calculated using the logistic regression method, which enables the analysis of the relationship between several independent variables (the values of the threshold PCR cycles for the studied DNA samples for each marker) and dependent one (DNA sample from tumor or normal tissue). As follows from the Figure 2 and Table 3, the tumor-marker combination significantly increases sensitivity and specificity of LC diagnosis: AUC value evaluates 0.911.
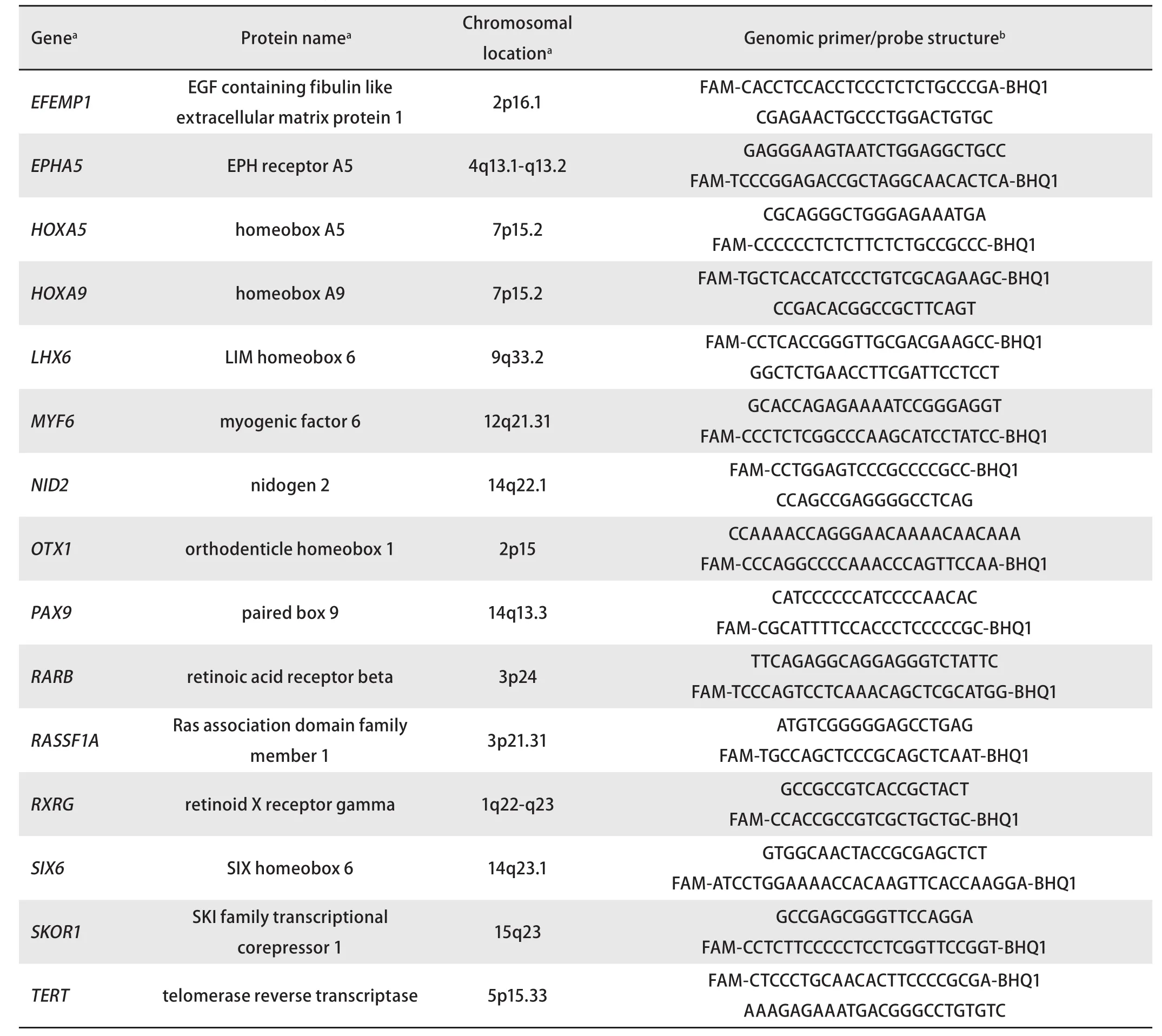
Tab 1 The genes selected for the study and a list of genomic primers and probes
Discussion
Nowadays, a large number of LC epigenetic determinants have been described which are characterized by the differences in DNA methylation status between tumor and normal lung tissue cells.
Thus Zhao Y et al. investigated 101 tumors and 30 normal lung tissue samples and demonstrated that regulatory regions of MYF6, SIX6 and RARB tumor-suppressor genes are the best epigenetic LC markers[12]. Another group investigated 103 tumors and 23 normal lung tissue samples and suggested using NID2 and RASSF1A genes as LC tumor-markers[13].Based on the analysis of DNA isolated from bronchial swabs of 333 LC patients and 323 healthy donors, Nikolaidis G.et al. identified a tumor-marker combination, including the regulatory regions of the TERT and RASSF1A genes,as the most informative for LC diagnosis[14]. Other authors characterized epigenetic LC markers within EFEMP1[15],EPHA5[16], HOXA5[17], HOXA9[18], LHX6[19], OTX1[20],PAX9[21], RXRG[22]и SKOR1[23]tumor-suppressor genes.
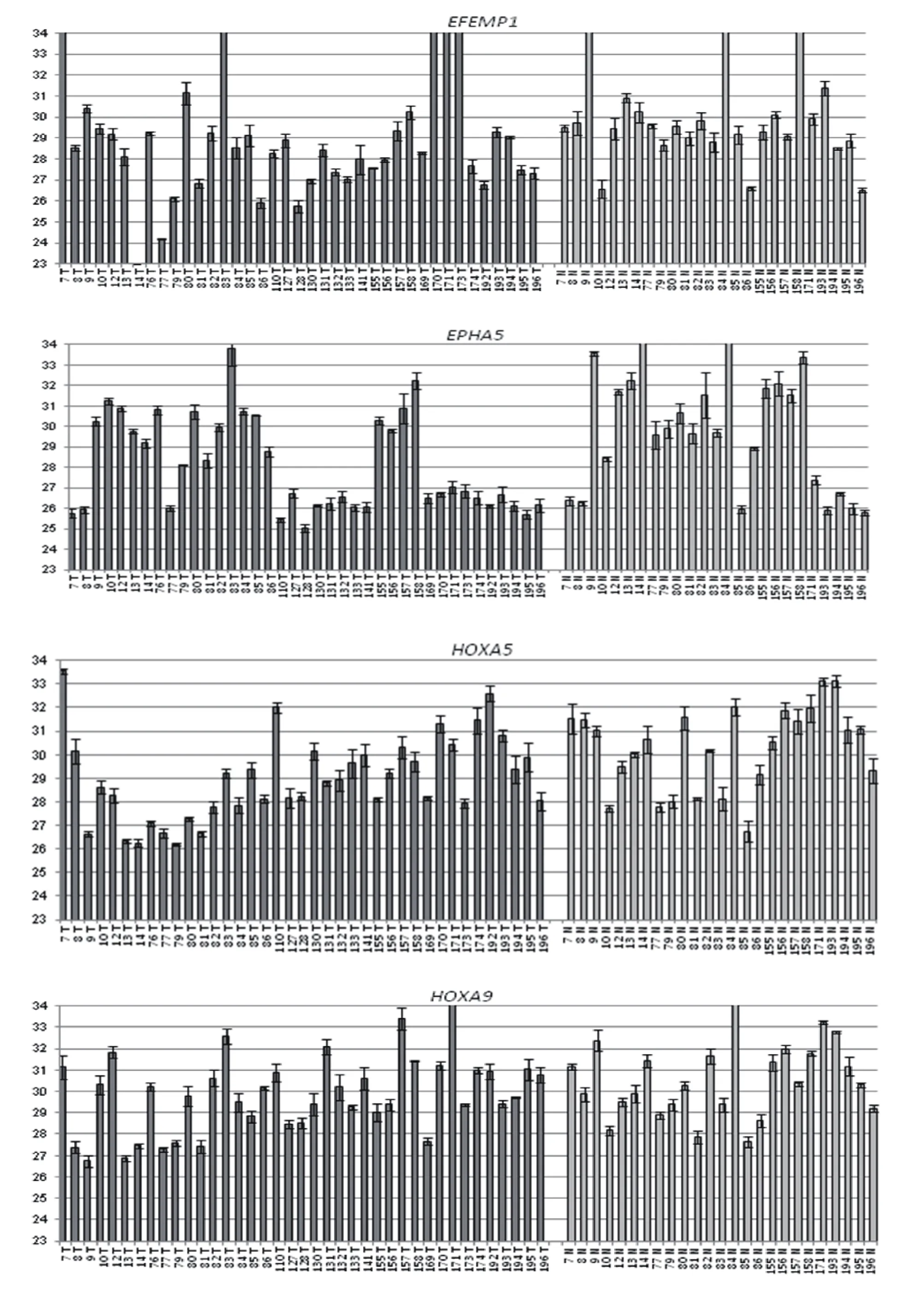

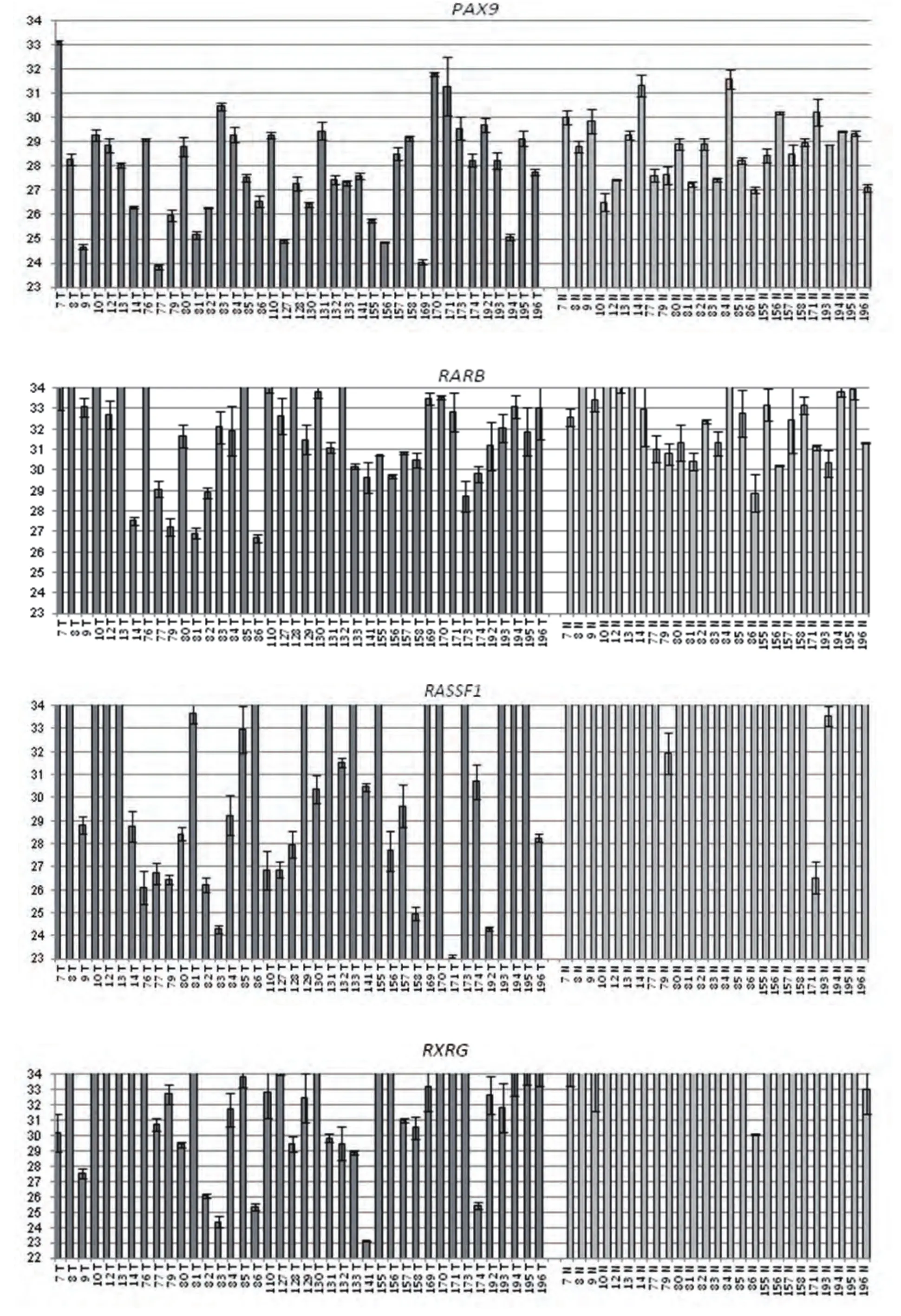
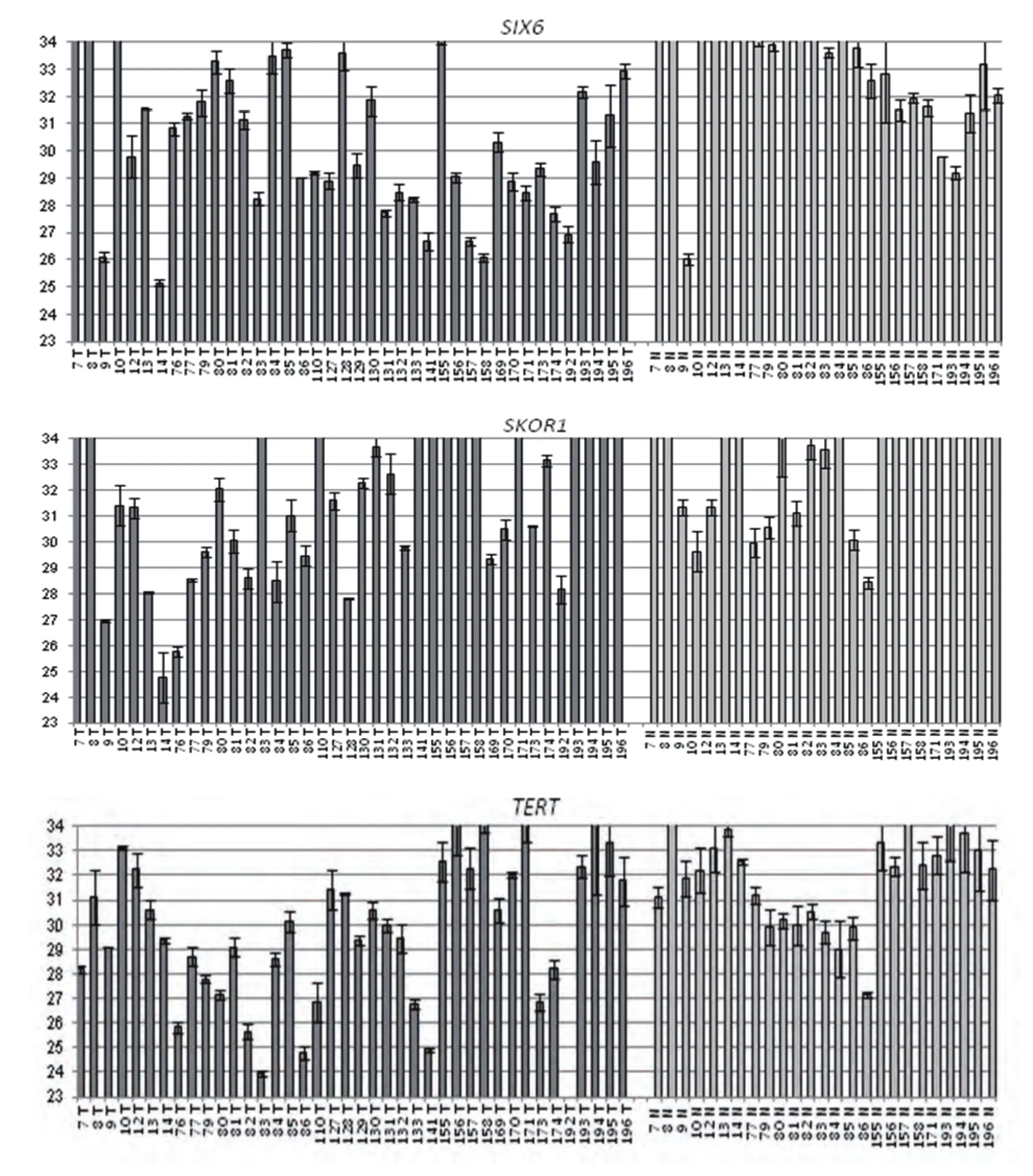
Fig 1 GLAD-PCR assay of selected RCGY sites. Cq values (with the standard deviation range) are given for DNA samples which are numerated below each diagram (T: tumor tissue, N: normal tissue).
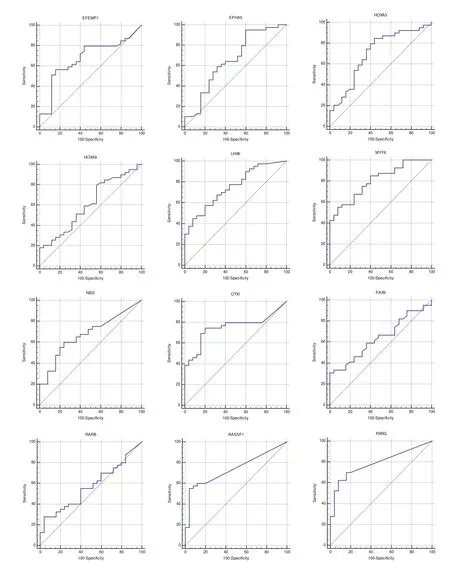
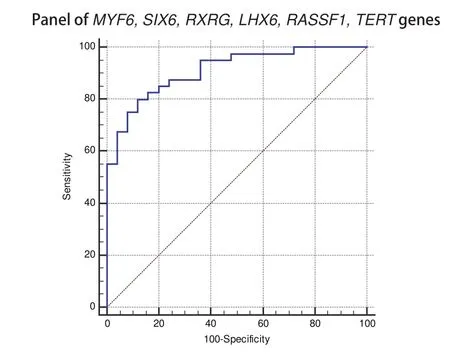
Fig 2 ROC-curves for GLAD-PCR analysis of selected RCGY sites in LC versus normal lung tissues. Resulting sensitivity, specificity, area under the ROC curve(AUC) with standard error and 95 % confidence interval (CI) are summarized in Table 3.
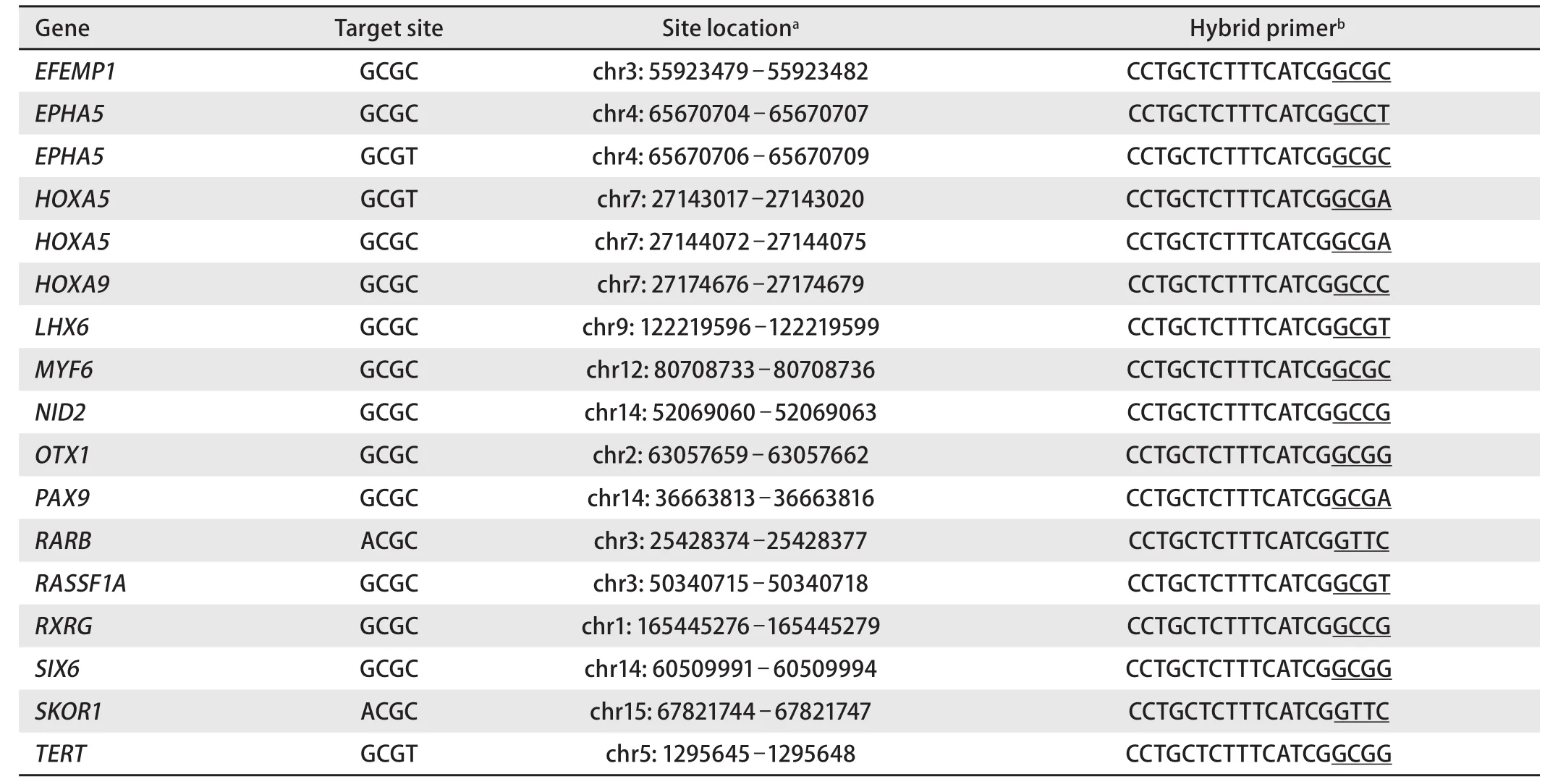
Tab 2 Studied genes with indication of the target RCGY site, its location and a structure of corresponding hybrid primer selected for GLAD-PCR assay
It is possible that such a variety of epigenetic markers is explained by the histological heterogeneity of LC, which is subdivided into two main types: small-cell lung carcinoma and non-small-cell lung carcinoma (about 15 % and 85 % of all cases, respectively). In turn, non-small cell LC includes three subtypes: squamous-cell carcinoma, adenocarcinoma and large-cell carcinoma, which occur in ratio close to 2.5:4:1[24].
GLAD-PCR opportunities for the diagnostics of lung cancer. The results obtained in this work demonstrate that GLAD-PCR analysis enables to determine R(5mC)GY sites in regulatory regions of tumor-suppressor genes in DNA samples isolated from LC tissue.
We have previously shown that GLAD-PCR assay can be used to analyze any complex DNA mixture, for example,DNA isolated from a patient’s blood containing nucleic acids both from tumor and normal cells (vascular cells, connectivetissue cells, etc.). The site methylation degree determines the threshold cycle value (Cq) in real-time PCR[7].
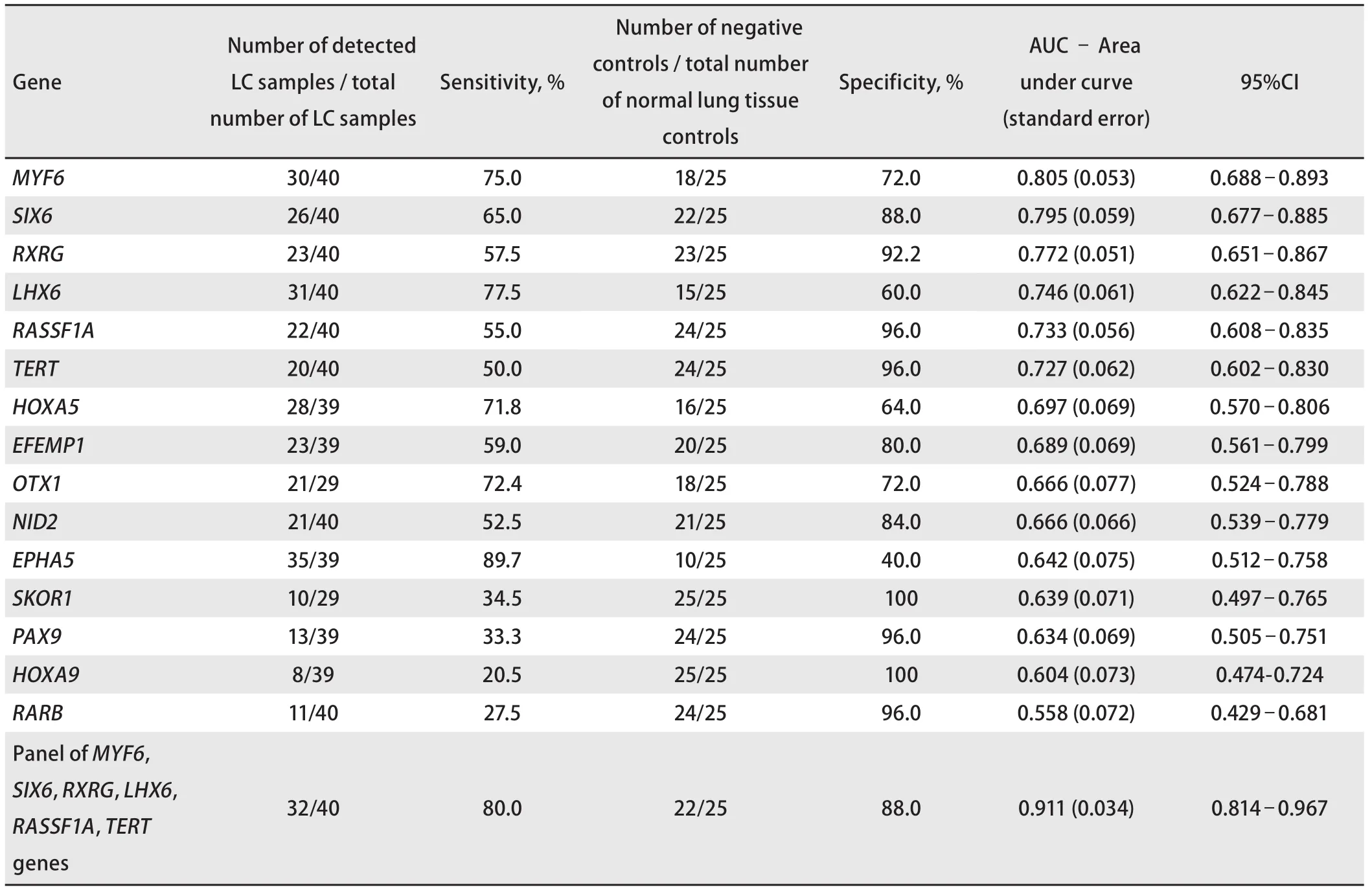
Tab 3 Receiver operating characteristics of GLAD-PCR assay of selected RCGY sites (sorted by AUC values) for LC versus normal lung tissue
The algorithm used for the ROC analysis of experimental data makes it possible to evaluate the reliability of the difference in the methylation status of tumor-suppressor genes in the DNA of tumor and normal cells and to identify potential epigenetic LC markers. The most promising markers (having maximum AUC values) are the target sites RCGY sites in regulatory regions of MYF6, SIX6, RXRG,LHX1, RASSF1A, and TERT genes.
A comprehensive study of gene panel significantly increases the sensitivity of the test (the total value is 80.0 %) in comparison with the analysis of individual markers (Figure 2 and Table 3).
Subsequently, in order to improve the diagnostic efficiency of the epigenetic LC marker panel, we intend to study a larger number of RCGY sites in other tumor-suppressor genes and a greater number of tissue samples belonging to different lung cancer subtypes. It will possibly change the panel composition.
Conclusion
GLAD-PCR assay enables to identify R(5mC)GY sites,arising from the aberrant methylation of regulatory regions of tumor-suppressor gene in LC tissue samples. In this study of DNA sample from clinical specimens we have shown a good diagnostic efficacy of the epigenetic LC markers panel including the RCGY sites in regulatory regions of MYF6,SIX6, RXRG, LHX1, RASSF1A and TERT genes: total sensitivity and specificity are 80.0 % and 88.0 %, respectively.The present study was supported by the Skolkovo Foundation (Under agreement NO. G102/16 06.12.2016 г.).Authors declare lack of the possible conflicts of interests.
