麻山药不同生长时期根际土壤微生物多样性及群落结构特征
2019-09-18康捷章淑艳韩韬孙志梅
康捷 章淑艳 韩韬 孙志梅
(1. 河北省微生物研究所,保定 071051;2. 河北农业大学,保定 071000)
根际土壤是指处于植物根际周围受根系生长直接影响的土壤,根际微生物是受植物影响最大的土壤微生物群体,与植物的生长发育关系密切[1]。根际微生物群落结构的变化对土壤中物质和能量的循环、有机质的分解与合成等方面产生重要影响[2]。研究表明,影响根际微生物群落结构变化因素有很多,不同植物间、同一植物的不同基因型,甚至同一植物的不同生长期其根际微生物的数量、种类也有很大差别[3]。根际分泌物为根际微生物的生长提供营养和能源物质,其对微生物的数量、种类、分布及微生物的生长都有一定的影响[4]。土壤微生物从各个方面影响着植物的生长发育,土壤微生物区系的动态平衡是保证土壤健康的重要标准,只有健康、良性循环的土壤生态环境才能保证植物的健康生长。有些根际微生物可以产生激素类物质促进土壤有机质分解、诱导植株增加抗性等,或者通过争夺营养、占领生态位等方式抑制病原菌的生长来间接促进植物的生长[5]。根际微生物群落结构失调,可能会导致病原菌数量的增加,从而使作物减产。因此研究不同作物土壤微生物的变化或同一作物不同生长时期土壤微生物的情况具有重要意义。
一直以来由于大多数的微生物在营养培养基中可培养性低或不能被培养的缺陷严重制约了微生物多样性的研究。随着分子生态学研究技术的发展,以 DNA 为基础的分子分析技术,如PCR-RFLP、DGGE/TGGE、SSCPP 等可以克服传统培养技术的限制,从分子水平来直接分析微生物群落结构的变化,然而这些技术存在检测限低、工作量大等缺陷[6]。高通量测序等非传统培养方法成为了研究土壤微生物多样性和群落结构特征的有力工具,其具有速度快,耗时少,成本低廉,准确度高等优点,能更真实地反映环境中群落的特点。目前,高通量测序技术主要是 Illumina、Roche454 和 Life Technologies 等公司开发的测序平台[7]。
张亮等[8]研究了不同品种百合在不同生育期根际土壤微生物种类、数量的变化规律,结果显示在生育期内,细菌数量最多;其次为真菌,放线菌。孙建波等[9]通过对香蕉不同生长期根际土壤细菌多样性和数量的研究,结果显示,细菌数量呈现先增加后减少的趋势,多样性逐渐减少。仝利红等[10]研究草莓不同生育期根区微生物多样性及动态变化,结果表明,根区可培养微生物总量从开始生长期到盛果期逐渐增多,细菌群落多样性指数和丰度在生长末期达到最高,而真菌在现蕾期达到最高。常颖萃等[11]测定了竹荪不同生育期内土壤中细菌、真菌、放线菌3种微生物的数量,研究了土壤微生物动态变化规律。胡延森等[12]对黄瓜根际主要微生物类群在不同生育期变化的研究,结果表明,根际微生物的数量一般是由栽种时开始升高,到花期或盛果期时达到最高峰,生长后期有下降趋势,同时黄瓜生长对某些种群数量分布有一定影响,也说明这些类群微生物可能是对黄瓜花期生长起特殊作用。
目前尚未有报道对麻山药不同生长时期根际土壤微生物多样性及群落结构特性的研究,本研究以麻山药为实验材料,采集麻山药苗期、花期和收获期的根际土样。运用 Illumina 测序平台对土壤样品细菌16S rDNA和真菌ITS rDNA 进行了高通量测序,通过比较不同时期麻山药根际土壤微生物多样性、均匀度和优势种群集中度的变化及真菌、细菌量的变化,探究3个不同生长期的麻山药根际土壤细菌群落结构多样性及变化,从而揭示麻山药不同生长期微生物群落特点及变化规律,这对评价生长期对土壤微生物多样性的影响具有重要意义。
1 材料与方法
1.1 材料
本研究所选地块为河北省蠡县小钟麻山药农业合作社的麻山药地,第2年种植。山药品种,紫药。播前土壤有机质含量7.12 g/kg、碱解氮28.45 mg/kg、速效磷6.73 mg/kg、速效钾63.37 mg/kg。测定方法参照文献[13]。
在2016年5月1日,7月18日(苗期),8月24日(花期)和10月15日(收获期)进行样品采集,在试验地内选取5个取样点,拨开麻山药根系周围的表土层,在离麻山药根基轴表面1-5 mm处,用抖落法获取其上黏附的土壤作为根际土壤[14]。不同时期的5个土样混合并除去较大根系等杂物后作为该时期土壤的一个样品,每组做3个平行,装于无菌牛皮纸袋中带回实验室。新鲜的土壤置-20℃冰箱冷冻保存,用于后续微生物多样性的测定。
1.2 方法
1.2.1 土壤DNA提取与检测 采用土壤基因组DNA提取试剂盒(离心柱型,天根生化科技有限公司)提取土壤总DNA,琼脂糖凝胶DNA回收试剂盒(普通离心柱型,天根生化科技有限公司)进行纯化,经1%的琼脂糖凝胶电泳检测及微量核酸蛋白检测仪检测浓度后置于-20℃冰箱保存备用。
1.2.2 基因扩增 PCR反应体系:DNA模板1 μL,10×Buffer 5 μL,2.5 mmol/L dNTPS 4 μL,Taq DNA聚合酶0.5 μL,上下游引物各1.5 μL,无菌超纯水补充至 50 μL。
以30-50 ng DNA为模板,使用金唯智设计的PCR引物扩增原核生物16S rDNA上包括 V3、V4 及V5的3个高度可变区。采用包含CCTACGGRRBGCASCAGKVRVGAAT序列的上游引物和包含GGACTACNVGGGTWTCTAATCC序列的下游引物扩增V3和V4区,采用包含GTGYCAGCMGCCGCGGTAA序列的上游引物和包含CTTGTGCGGKCCCCCGYCAATTC 序列的下游引物扩增V4和V5区。以5-50 ng DNA 为模板,PCR扩增真菌ITS rDNA上的 ITS2可变区。
扩增程序:94℃预变性5 min;94℃变性50 s,50-60℃退火50 s,72℃延伸1.5 min,30个循环;72℃延伸10 min。用1%的琼脂糖凝胶电泳检测PCR扩增产物。
1.2.3 高通量测序 高通量测序文库的构建和基于I11umina MiSeq平台的测序由GENEWIZ公司(Suzhou,China)完成。双端测序得到的正反向reads 首先进行两两组装连接,过滤拼接结果中含有N的序列,保留序列长度大于200 bp的序列。经过质量过滤,去除嵌合体序列,最终得到的序列用于OTU分析,使用 VSEARCH(1.9.6)进行序列聚类(序列相似性设为97%),比对的16S rRNA参考数据库是Silva 119,ITS rRNA参考数据库是UNITE ITS database(https://unite.ut.ee/)。 然 后 用 RDP classifier(Ribosomal databaseprogram)贝叶斯算法对OTU的代表性序列进行物种分类学分析,并在不同物种分类水平下统计每个样本的群落组成。基于OTU的分析结果,采用对样本序列进行随机抽样的方法,分别计算Shannon、Chao1等α多样性指数,并作出稀释曲线。通过层次聚类(Hierarchical clustering)中的非加权组平均法构建(Unweighted pair group method with arithmetic mean,UPGMA)进化树。
2 结果
2.1 根际土壤细菌群落结构分析
2.1.1 麻山药不同生长期土壤样本细菌测序特性 由于高通量测序常常会出现一些突变等错误,对测序结果的原始数据进行优化处理,处理步骤及参数:将2条序列进行比对,根据比对的末端重叠区进行拼接,拼接时保证至少有20 bp的重叠区,去除拼接结果中含有N的序列;去除引物和接头序列,去除两端质量值低于20的碱基,去除长度小200 bp的序列;将上面拼接过滤后的序列与数据库进行比对,去除其中的嵌合体序列(Chimera sequence),得到最终的有效数据。
对于细菌群落来说,样品在不用生长时期统计结果如表1所示。
随着样品量的加大,可能出现测序检测到的物种种类随之增加的状况,稀释曲线(Rarefaction curve)是调查样品的物种组成和预测样品中物种丰度的有效工具,在生物多样性和群落调查中,常用于判断样品量是否充分及估计物种的丰富度。从图1和图2可以看出,麻山药不同生长期样品对应的稀释曲线均基本趋于平缓,说明取样基本合理,细菌(或真菌)群落结构的置信度较高,能比较真实地反映出样品在不同生长时期的细菌(或真菌)群落。
2.1.2 麻山药不同生长期土壤细菌多样性指数分析 群落生态学中,α多样性主要关注单样本的多样性分析,可以反映微生物群落中物种的数目,通过一系列统计学指数的分析来估计环境群落的物种丰度和多样性。由表2可以看出,不同生长期细菌的Good's Coverage指数都接近100%,说明样本序列中没有被测出的概率很低,在该水平上的测序结果能够反映出所测样本中细菌的真实情况。计算菌群丰度的指数有ACE和Chao,计算菌群多样性指数有Shannon和Simpson,由表2可以看出,不同生长期土壤中细菌丰度和多样性之间没有显著性差异。
2.1.3 不同生长期细菌种类分布 使用RDP classifier对97%相似水平的OTU代表序列进行分类学分析,从而获得每个样品在各个水平对应的群落组成。如表3所示,不同时期的土壤样品中检测到12个以上细菌门,其相对丰度均大于1%,说明这12个门的细菌在本实验中所取的土壤细菌群落结构组成中占主要地位。

图2 不同时期土壤样品真菌稀释曲线

表2 不同时期土壤细菌的α-多样性指数

表3 主要细菌门的相对丰度
2.1.4 不同生长期主要细菌物种分布 与Silva数据库相对比,获得各样品细菌在门水平下Top30的物种分布图。如图3所示,结合上表分析得出,不同生长期土壤中变形菌门均为最优菌群,苗期、花期和收获期所占的比例分别达到35.22%、29.44%和28.30%,随着种植时间的延长,其所占的比例呈下降的趋势,即在麻山药苗期所占的比例最高,较其他两组都有显著性差异(P<0.05),花期和收获期两组之间没有显著性差异;其次为酸杆菌门,两者之和在不同生长期所占的比例之和超过了50%,苗期、花期和收获期酸杆菌门所占的比例分别为21.03%、24.85%和23.28%,在麻山药花期土壤中酸杆菌门达到最高。此外,放线菌门、芽单胞菌门和拟杆菌门在麻山药种植中不同生长期内均为优势菌群(所占比例超过5%),其中拟杆菌门同样在麻山药苗期达到最高,为9.15%,比花期和收获期高3.69和3.75个百分点,有显著性差异(P<0.05)。其他菌群(所占比例超过1%,但低于5%)中,硝化螺细菌门所占比例在花期接近5%,显著高于其他两个时期,而绿弯菌门和浮霉菌门均在收获期所占比例最高。
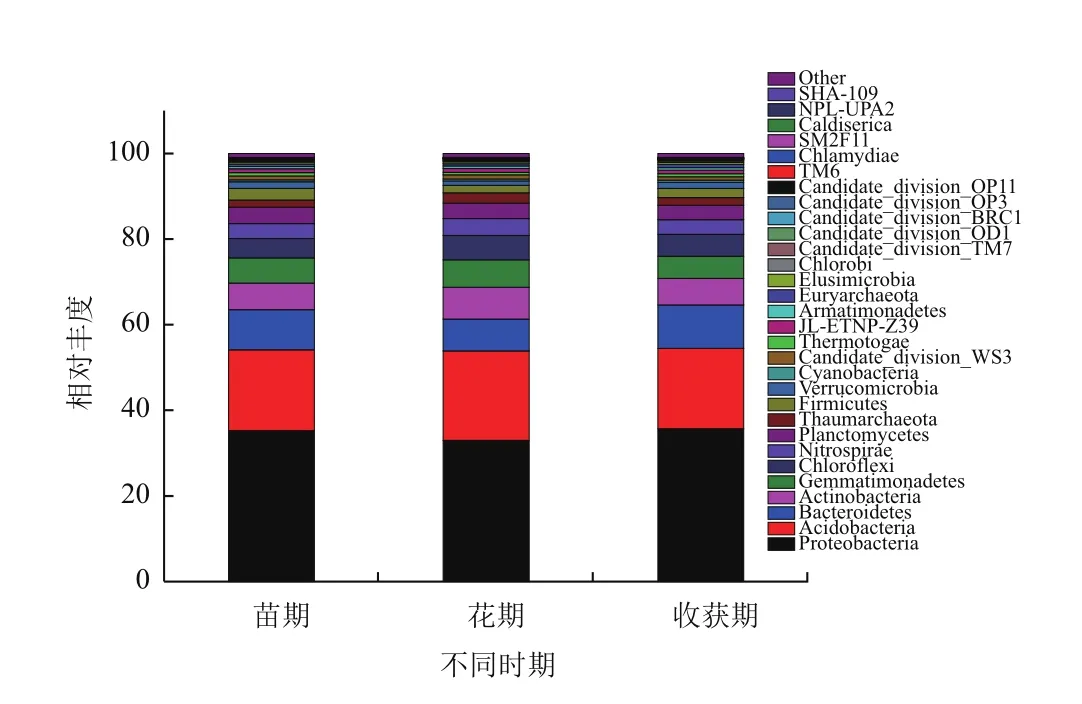
图3 不同时期土壤样品在门水平前30的细菌物种分布图
2.1.5 不同生长期细菌群落相似度聚类树分析 样品聚类分析利用各样品序列间的进化信息来比较在特定的进化谱系中是否具有显著的微生物群落差异,使用非加权组平均法(Unweighted pair-group method with arithmetic means,UPGMA)将样品进行聚类,结果(图4)显示,不同生长时期土壤中细菌组成可以分为两大类,图4中苗期和花期土壤的细菌群落组成具有很高的相似性,苗期第1组和第3组具有很高的相似性,并与第2组的土壤样品聚为一类,花期第3组和第2组具有很高的相似性,并与收获期第一组的土壤样品聚为一类,同时这6组土壤样品的趋同性较高,而收获期第3组和第2组样品具有很高的相似性,并与花期第1组的土壤样品聚为一类。由此可以得知,麻山药不同生长时期的土壤细菌群落多样性之间虽然有不同程度的差异,但是苗期和花期两组之间群落组成结构相似性较高,与收获期之间也存在趋同性。
2.2 根际土壤真菌群落结构分析
2.2.1 麻山药不同生长期土壤样本真菌测序特性 对真菌群落来说,样品在不同生长时期过滤后统计结果如表4所示。

图4 细菌群落结构UPGMA聚类图

表4 不同时期真菌样本统计数据
2.2.2 麻山药不同生长期土壤真菌多样性指数分析 由表5看出,不同生长期真的Good’s Coverage指数都接近100%,说明样本序列中未被测出的概率很低,在该水平上的测序结果能够反映出所测样本中真菌的真实情况。麻山药不同生长期土壤中真菌丰度之间同细菌一样无显著性差异,但表现为收获期>花期>苗期。由表5中Shannon和Simpson指数,同样真菌多样性也表现为收获期>花期>苗期,且收获期和花期的多样性指数显著高于苗期。由此可见,麻山药种植过程中,不同生长期内真菌变化较大,随着种植时间的延长,真菌丰度和多样性明显提高,说明真菌数量也有明显提高。同时,在不同生长发育时期,麻山药根系生理活动代谢活动的强弱也不同,生长后期会出现特定菌属所占比例显著高于其他时期,这与作物生长所需有关。
2.2.3 不同生长期真菌种类分布 同样使用RDP classifier对97%相似水平的OTU代表序列进行分类学分析,获得每个样品在各个水平对应的群落组成。如表6所示,不同时期的土壤样品中检测到13个以上真菌属。可见,这13个属的真菌在本实验中所取的土壤真菌群落结构组成中占主要地位。
2.2.4 不同生长期主要真菌物种分布 对比Silva数据库,各样品真菌在属水平下Top30的物种分布图如图5所示。结合上表分析得出,与细菌不同的是不同生长期土壤中最优真菌菌属不同,放射毛霉属是麻山药苗期最优菌属,所占比例为13.8%,而花期和收获期的最优菌属为被孢霉属,所占比例分别为6.97%和7.86%。根霉属在麻山药种植初期,即苗期所占的比例最高,达2.51%,比花期和收获期高2.11和1.89个百分点,差异显著(P<0.05)。肉坐菌属、毛壳菌属和绿僵菌属,在麻山药的花期所占比例最高,分别为2.47%、1.16%和1.02%,而其他两个时期的比例却只有不到1.0%甚至更低,有显著性差异(P<0.05),毛壳菌属作为生防菌在防治植物病害方面最为突出,是腐生子囊菌中数量最多和意义较大的类群之一。被孢霉属、接合菌属、翅孢壳属和球囊霉属所占的比例在麻山药种植期间呈上升趋势,即在收获期最高,分别达到7.86%、1.25%、1.26%和1.12%,显著高于其他两个时期,花期和收获期的比例均不到0.5%(被孢霉属除外)。
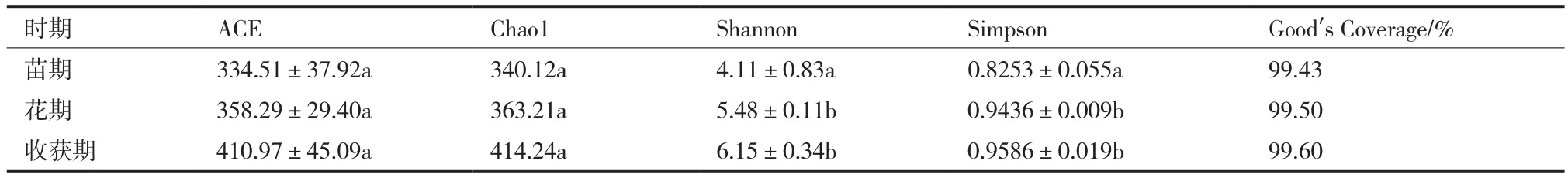
表5 各处理组土壤真菌的α-多样性指数
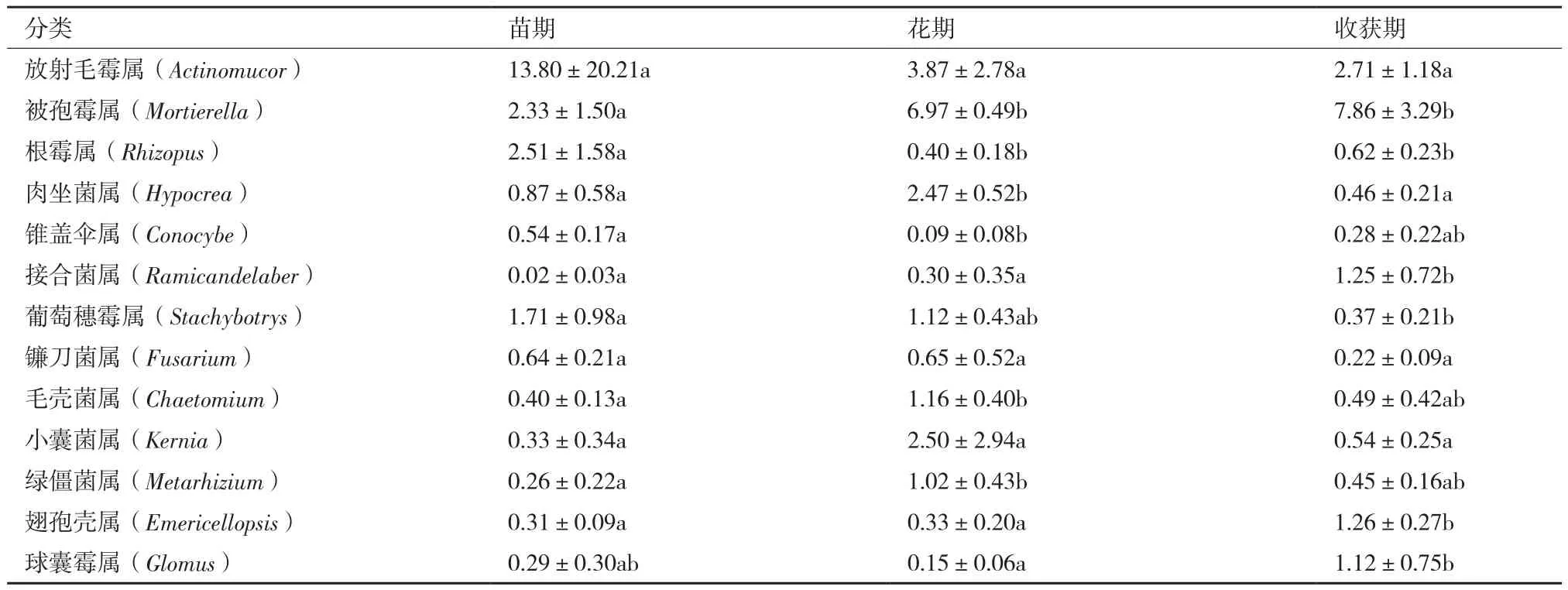
表6 主要真菌属的相对丰度
2.2.5 不同生长期真菌群落相似度聚类树分析 不同生长时期麻山药土壤样品真菌群落结构聚类,结果(图6)显示,不同时期土壤中真菌组成分为两大类,花期第3组土壤样品与第2组之间具有很高的相似性,并与苗期第3组土壤样品聚为一类;收获期第2组土壤样品和第3组之间有很高的相似性,并与第1组土壤样品聚为一类,这6组样品与苗期第2组样品之间有较高的相似性,并与花期第一组样品聚为一类;苗期第1组土壤样品单独聚为一类,与其余几组之间真菌群落结构存在相对较大的差异性。
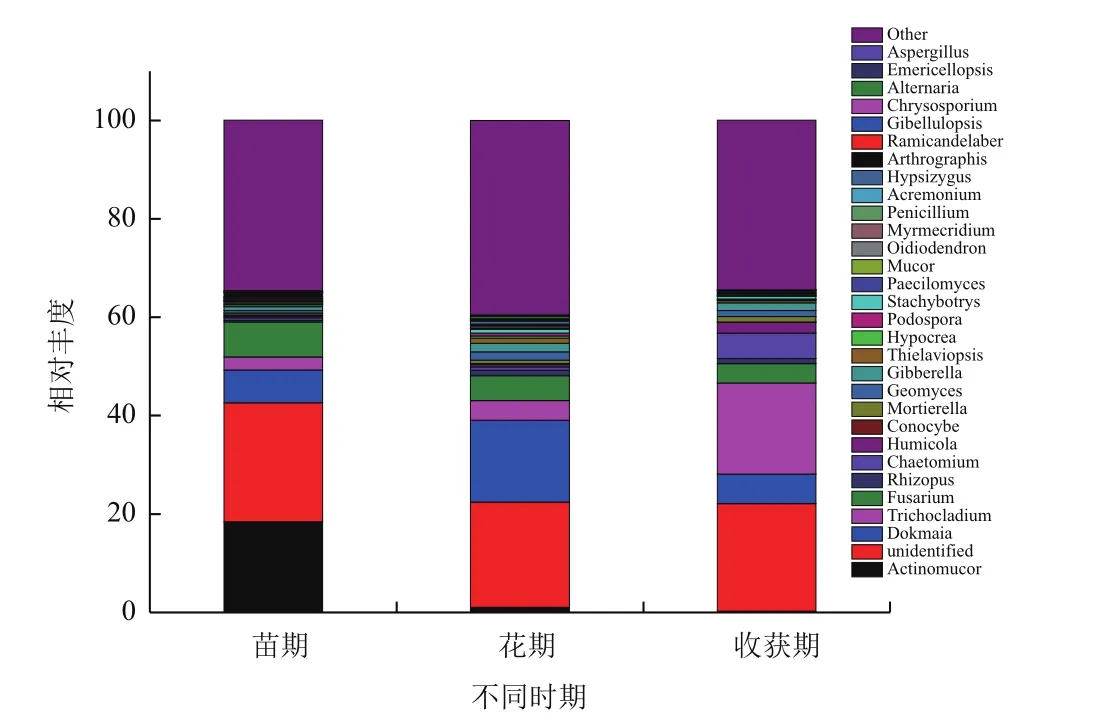
图5 不同时期土壤样品在属水平前30的真菌物种分布图
3 讨论
土壤微生物区系是土壤质量的变化的主要因素之一,同时也决定了土壤是否健康,土壤微生物区系与许多土传病害之间都有着密切的联系,研究表明,土壤健康的显著特征是土壤微生物生物量大、多样性高和土壤微生物区系组成的动态平衡[15]。微生物培养计数作为传统的分析方法在土壤微生物研究中具有重要作用,但是,传统的分析方法不能反映其在土壤中的真实情况,需要采用新的方法进行研究分析[16]。随着人们对微生物在农业生产中重要作用的认识不断加深,用土壤微生物学特性来评价土壤的健康程度和质量日渐被认可[17],用高通量手段测定的多样性指标可以作为评价土壤健康程度的一种指标,与传统的培养测数法相比,都是一种手段。

图6 真菌群落结构UPGMA聚类图
本研究运用高通量测序技术探究3个不同生长时期麻山药根际土壤中细菌和真菌群落结构变化,计算菌群丰度的指数有ACE和Chao,由Chao于1984最早提出[18],在生态学中常用来估计物种总数,计算菌群多样性的指数有Shannon和Simpson,辛普森(Simpson)多样性指数,由Edward Hugh Simpson于1949提出,在生态学中常用来定量的描述一个区域的生物多样性,其值越高,说明群落多样性越高[19]。通过对麻山药不同生长期根际土壤分析可知,不同生长期土壤中细菌丰度和多样性之间没有显著性差异,变形菌门和酸杆菌门是麻山药根际细菌群落的最优菌群。而真菌在不同生长时期变化趋势明显,从开始生长期到收获期逐渐增多,在收获期时多样性指数和丰度达到最高,放射毛霉属和被孢霉属始终是真菌群落的最优菌群。结合其他优势菌群在不同生长时期的明显变化,可以看出,这些菌群跟麻山药生长发育密切相关。这表明麻山药的生长期对根际土壤细菌和真菌群落结构均有重要的影响。
有研究表明,真菌是参与土壤有机物质分解过程中的主要成员之一,它们分解纤维素、半纤维素、木质素、单宁等化合物的能力远大于细菌,从而有利于改良土壤结构,提高土壤肥力,促进作物生长[20]。研究显示真菌数量在收获期达到最高值,表明麻山药种植后期根际生理代谢活动加强,其产生的代谢产物仅有利于某些特定菌群的生长,从而显著高于其他时期,而这些优势菌群对其他菌群具有竞争优势,结构造成部分真菌成为优势菌群并大量繁殖,另外一些菌群则在竞争中被抑制。早在1954年,Martin和Moorc[21]发现,燕麦种子被球毛壳和螺卷毛壳侵染后,会使谷物、大麦等作物的幼苗免受镰刀菌的再侵染[22],这点与测定结果相一致,被孢霉属、接合菌属、翅孢壳属和球囊霉属所占的比例在麻山药种植期间呈上升趋势,在收获期最高,说明这几种真菌在麻山药种植后期发挥作用。麻山药种植不同时期内真菌含量及所占的比例各不相同,说明不同类别的菌属在不同时期所起的作用不同,这跟作物生长所需有关。
研究同时发现,麻山药不同生长时期所有样品中共包含29个细菌门,其中变形菌门和酸杆菌门的相对丰度最高,两者在不同时期所占比例均超过了50%;其次是放线菌门、芽单胞菌门和拟杆菌门,这3大门类分别所占的比例均超过了5%。袁红朝等[23]分析稻田土壤细菌的群落结构,发现变形菌、酸杆菌和绿弯菌是优势细菌;其次是放线菌。牛世全等[24]分析河西走廊地区盐碱土壤群落结构,发现变形菌门含量最高,占 28.56%;其次是放线菌门,占 17.2%。杨菁等[25]发现降香黄檀不同混交林土壤细菌多样性主要菌群有变形菌门、酸杆菌门、放线菌门及绿弯菌门。结果均与本研究相一致。而真菌相对丰度在不同时期变化显著,苗期、花期和收获期分别包含有64、67和75个真菌属,其中放射毛霉属和被孢霉属是真菌群落的最优菌群,两者在不同时期所占的比例约15%,结合其他优势菌群在不同生长时期的明显变化,可以看出,这些菌群在不同生长时期所占的比例有所不同,可能与作物生长发育有关。
4 结论
本文研究了麻山药在苗期、花期和收获期根际土壤微生物群落结构的变化,变形菌门和酸杆菌门是麻山药根际细菌群落的最优菌群,放射毛霉属和被孢霉属是真菌群落的最优菌群,同时真菌数量在收获期达到最高值,可以看出,这些菌群含量不同与麻山药生长发育密切相关。因此,为后续在研究麻山药土传病害的方面提供理论依据,可通过研发土壤菌剂调节剂的手段,从而改变土壤微生物菌群的组成,使其通过细菌与细菌之间、真菌与真菌之间或细菌与真菌之间的相互作用,达到抑制病原微生物的生长,提高抗病性的目的。
