基于HeteSim的疾病关联长非编码RNA预测
2019-09-16郭杏莉孙宇彤苑倩倩
马 毅 郭杏莉 孙宇彤 苑倩倩 任 阳 段 然 高 琳
(西安电子科技大学计算机科学与技术学院 西安 710071)
随着已确定的lncRNA的数量持续增长,许多相关的数据库、计算方法被提出来,其中包括通用的数据库GENCODE[6],针对lncRNA的专用数据库lncRNAdb[7],LncRbase[8],LncRNA2Function[9],LncRNA2Target[10],同时包括基于网络的大规模lncRNA功能预测方法lncGFP[11],以及通用的计算模型和框架[12].关于lncRNA在普通疾病和癌症中的作用,分别有LncRNADisease[13]和Lnc2Cancer[14]数据库.即使有一定数量的lncRNA-疾病关联关系已经得到实验验证,不可忽略的是,绝大多数lncRNA-疾病关联关系仍然是未知的.因此,分析lncRNA与疾病关联关系并预测潜在的关联关系具有重要的研究价值和社会意义.这些研究不仅可以帮助我们加深对复杂疾病在分子层面的致病机理的理解,而且可以利用lncRNA作为疾病诊断、预测的生物靶标以及治疗和预防的药物靶标.
预测潜在的疾病与lncRNA关联关系的计算方法可分为2大类:基于机器学习和基于网络的方法.基于机器学习的方法通常使用疾病与lncRNA关联关系来训练学习模型,然后用学习得到的模型来预测新的关联关系.这类方法整合了各种生物信息来注释lncRNA.例如,Zhao等人[15]使用朴素贝叶斯模型来整合基因组、调节子和转录组特征,进而识别与癌症相关的潜在lncRNA.这个方法需要阴性的训练样本(即与疾病无关的lncRNA)来训练模型,考虑到并没有这种实验验证的阴性样本,在这项研究中,所有未知的lncRNA-疾病关联关系被认为是阴性样本用于训练.最近,一个半监督模型——正则化最小二乘(RLS)[16]克服了这一限制,该模型不需要阴性的训练样本.
相对于比较少的基于机器学习方法的研究,许多基于网络的方法被提出来预测与疾病相关的潜在lncRNA.基于网络的方法通常根据lncRNA与疾病的关联得分大小对候选的lncRNA进行排序,进而预测致病基因.最常用的算法是标签传播算法,比如随机漫步(RWR)[17-21]和KATZ[22].这些研究的主要区别在于传播算法所应用的底层网络不同.例如:Sun等人[17]将RWR应用于lncRNA功能相似网络(lncRNA FSN);Liu等人[18]基于lncRNA和蛋白质编码基因表达谱构建了蛋白质编码基因-lncRNA二部网络,然后利用RWR算法来预测癌症相关的lncRNA;与此同时,Zhou等人[19]和Ganegoda等人[20]结合lncRNA相似网络建立了lncRNA-疾病异质信息网络,然后在该网络上应用RWR算法预测潜在疾病lncRNA关联关系.这些基于网络的方法是基于一种观察结果提出的,即在功能上类似的lncRNA通常与相同或相似的疾病联系在一起,即疾病模块原理.以上方法都是通过构建网络提出基于网络的计算模型,有的方法结合基因表达谱数据等构建网络,所构建网络结合了多种信息的逻辑关联网络,构建方法相对复杂.
本文使用了一种异质信息网络中节点相关性计算方法——HeteSim,该方法用来预测基因和疾病的关联关系,得到了很好的实验验证[23].因此,我们将这种方法应用到lncRNA-疾病异质信息网络中,通过挖掘网络中疾病与lncRNA之间的关联关系,计算疾病与lncRNA关联得分,预测潜在疾病关联lncRNA,预测结果优于其他方法.
1 算 法
1.1 异质信息网络构建
预测lncRNA与疾病之间的关联关系可以理解为lncRNA-疾病异质信息网络上的一个相关性搜索任务.异质信息网络是一种特殊的信息网络,下面是信息网络的定义,在此基础上可以定义得到同质信息网络和异质信息网络.
定义1.信息网络.给定一个模式S=(A,R),它由对象类型集合A和关系集合R构成.信息网络被抽象定义为一个有向图G= (V,E),其中,V是所有实体节点的集合,E是所有关系边的集合.并且存在一个节点类型的映射函数φ:V→A和一个边类型的映射函数θ:E→R,对于每个对象v∈V属于一种特殊的对象类型φ(v)∈A,每个链接e∈E属于一种特殊的关系类型θ(e)∈R,那么这种网络类型就是信息网络.当对象类型的种类|A|>1或者关系类型的种类|R|>1时,这种信息网络是异质信息网络.例如图1(a)就是由电影数据构建成电影异质信息网络.
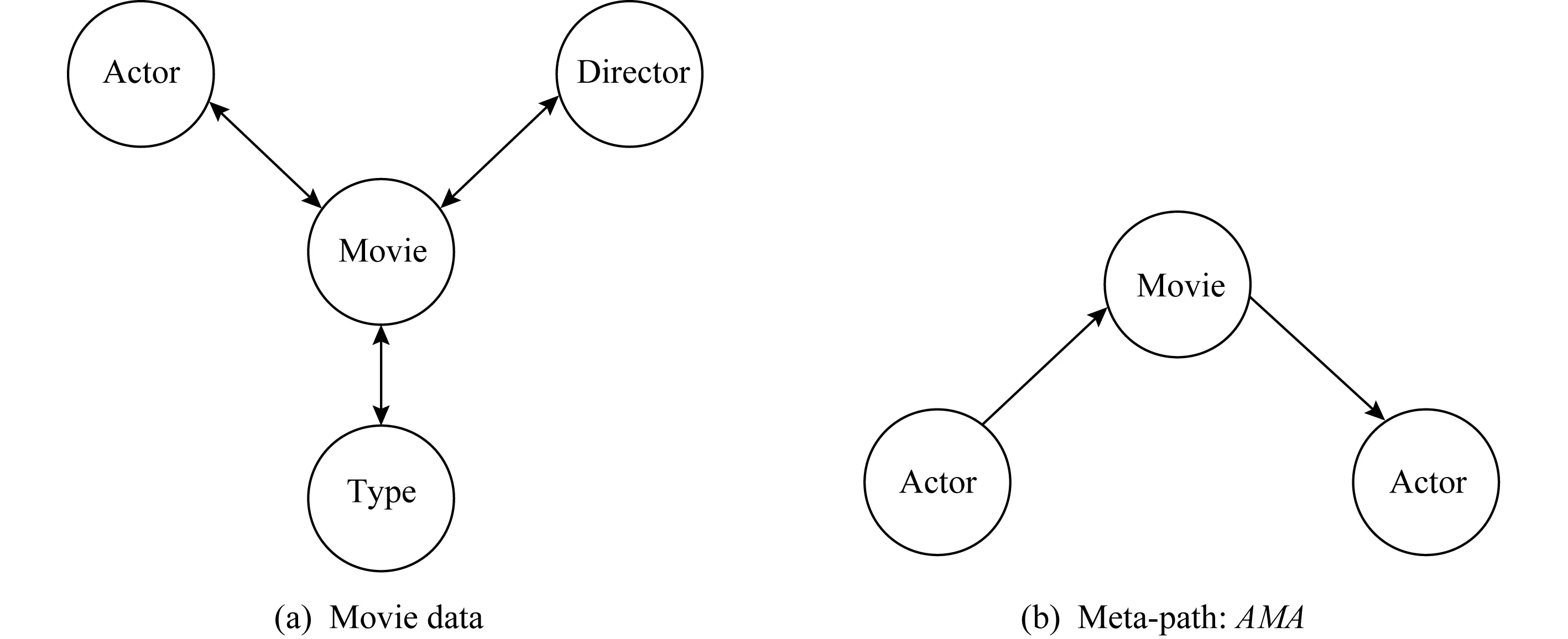
Fig. 1 Heterogeneous information network instance and meta-path[24]图1 异质信息网络实例和元路径[24]
基于已知的lncRNA与疾病关联关系,构建lncRNA-疾病异质信息网络,如图2(a)所示.网络中包含2种类型节点,分别为lncRNA和疾病,包含1种类型的边,即lncRNA-疾病关联关系.为了集成更多的疾病相关的基因信息,类似地,我们集成了OMIM(online mendelian inheritance in man)数据库中已知的编码基因与疾病的关联关系,将上面所构建的异质信息网络进行了扩展.扩展后的网络中包含2种类型节点,分别为基因和疾病,其中基因包括lncRNA和从OMIM中集成的编码基因.相应的边扩展为基因-疾病关联关系.lncRNA与疾病的关联预测在基因-疾病关联异质信息网络上进行.
1.2 元路径选择
由于HeteSim是一种路径约束的相关性计算方法,所以选择相关路径是非常重要的.构建了异质信息网络之后,我们的目的是要研究lncRNA和疾病的相关关系,即通过现有的异质信息网络预测出lncRNA是否和其他疾病相关联,因此我们选择lncRNA-疾病-lncRNA-疾病(LDLD)作为元路径,如图2所示.在此路径下使用HeteSim算法计算lncRNA和疾病之间的相关性,就能根据已有的关系预测出潜在的lncRNA-疾病关联关系.
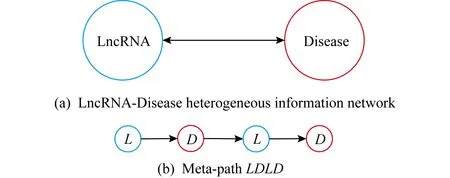
Fig. 2 LncRNA-Disease heterogeneous information network and meta-path LDLD图2 LncRNA-疾病异质信息网络和元路径LDLD
1.3 模型描述
石川等人[24]提出了HeteSim算法来计算异质信息网络中任意节点对的相关性,该方法具有对称特性而且可以计算相同或不同类型对象之间的相关性,从而适用于很多的应用.HeteSim是一种基于双向随机游走(pair-wise random walk)的相关性计算方法,它将元路径P分割成2条相等长度的元路径PL和PR,之后将对象s和t分别沿着元路径PL和PR进行随机游走,最后将2个对象走到相同中间节点的概率作为s和t的相关性.
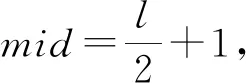
对于我们选择的元路径lncRNA-疾病-lncRNA-疾病(LDLD),由于路径长度是奇数,元路径两端的2个节点始终都不会在1个点相遇,因此我们需要插入中间类型M从而使路径可以等分成路径PL=LDM和PR=MLD,如图3所示:
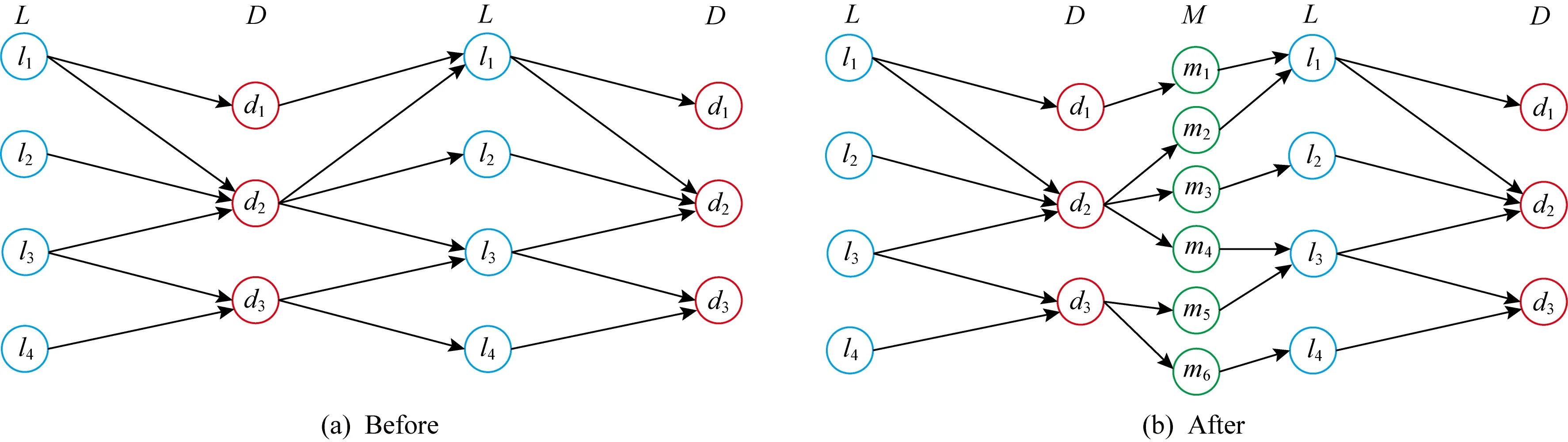
Fig. 3 Before and after insertion of the intermediate type M图3 插入中间类型M前后
下面介绍如何利用矩阵乘法计算lncRNA和疾病之间的关联得分.首先,我们定义2类矩阵:转移概率矩阵和可达概率矩阵.
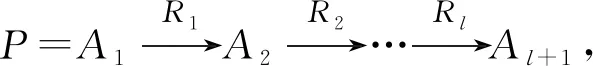
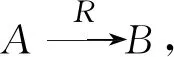
根据HeteSim的定义,类型L中的节点基于元路径P=LDLD到类型D中的节点之间的相似度为类型L的节点和类型D中的节点随机游走恰好在元路径中间类型M相遇的概率,计算公式为
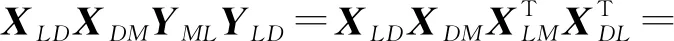
(1)
式(1)表明L和D之间基于路径P的相关性是2个概率分布的内积.
对于lncRNA和疾病类型中具体的对象l,d,基于路径P的关联得分计算为
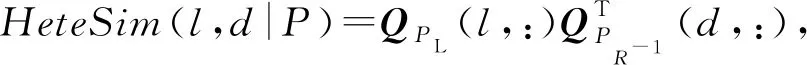
(2)
其中QP(l,:)为矩阵QP中对象l所对应的行向量.
为了使得HeteSim得分取值位于区间[0,1],还需要对计算出的关联得分进行标准化处理:

(3)
由式(1)~(3)我们就可以计算出lncRNA和疾病之间的关联得分.可以看到,计算HeteSim得分的过程主要包括3个部分:邻接矩阵标准化运算、矩阵连乘运算、相似度标准化运算.
2 实验结果与分析
2.1 实验数据
实验中所使用疾病与基因关联数据均来自文献[25],包括lncRNA与疾病关联数据以及已知的编码基因与疾病关联数据.lncRNA与疾病关联数据包括2个部分:1)来自LncRNADisease数据库[13]的数据,其中包含480条实验验证的lncRNA与疾病关联关系,涉及到166种疾病和118种lncRNA;2)在PubMed上进行文本挖掘得到的lncRNA与疾病关联数据,其中包含380条lncRNA-疾病关联的数据,包括226种lncRNA和145种疾病.
整合上述2种数据集,最终得到了578条lncRNA-疾病关联关系,其中包括295种lncRNA和214种疾病,构成了lncRNA-疾病异质信息网络.
编码基因与疾病关联数据来自OMIM数据库[26].针对上述lncRNA-疾病关联数据中涉及到的214种疾病,其中160种疾病可通过MIM编号在OMIM数据库中找到该疾病的致病基因,Yang等人[25]提取了OMIM数据库中这160种疾病与编码基因的关联关系,得到980条编码基因与疾病关联的数据条目,包括801个编码基因和160种疾病.
通过整合上述lncRNA与疾病关联数据、编码基因与疾病关联数据,得到1 558条编码-长非编码基因与疾病的关联关系,其中包括214种疾病和1 096种基因(编码基因或lncRNA),根据以上数据构建基因-疾病异质信息网络.
上述2个网络中的具体信息如表1所示:
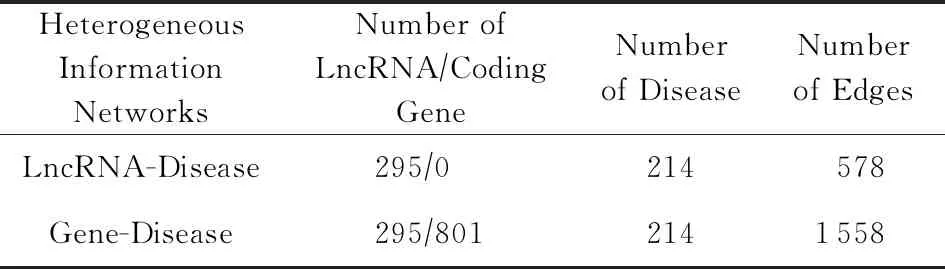
Table 1 Specific Information in the LncRNA/Gene-DiseaseHeterogeneous Information Network表1 lncRNA/基因-疾病异质信息网络中的具体信息
2.2 性能分析
对基因-疾病异质信息网络中不存在连边的基因与疾病对,采用HeteSim算法计算疾病与基因之间的关联得分,预测潜在的lncRNA和疾病关联关系.对每一个疾病,选取关联得分在top10的基因认为是其潜在的致病基因.
HeteSim在lncRNA-疾病异质信息网络中的性能通过留一交叉验证(leave-one-out cross valida-tion, LOOCV)实验来评估.由于二部网络中度为1的节点所关联边被移除后会成为孤立节点,不能通过网络方法和计算模型得到任何信息,因此本文的预测方法无法计算这些边的得分值.所以,在进行留一交叉验证之前应过滤这类边.最后,我们保留了532条边,其中包括103个疾病和163个基因(包括44个lncRNA和119个编码基因).对于保留的每一条关联关系中的疾病,我们在没有边相连的lncRNA中随机选取1个lncRNA与该疾病相连,构造本文实验的负样本.
在每次留一交叉验证运行过程中,我们删除1个已知的lncRNA-疾病关联边,然后在剩下的网络中应用HeteSim算法计算出删除边的HeteSim关联得分.这个被删除的边被认为是测试样本,剩下的网络结构被认为是训练样本.通过设定不同的阈值(topk%,1≤k≤100),我们使用ROC曲线和ROC曲线下的区域(AUC)来评估HeteSim在网络上的表现.ROC曲线的横轴是“假阳性率”(FPR),它是实际负样本中错误地识别为正样本的比例;纵轴是“真阳性率”(TPR),它是所有实际正样本中正确识别的正样本的比例.二者的计算公式为
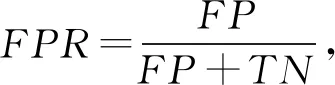
(4)

(5)
TPR表示的是移除的关联边排名在k%以内的比率;FPR表示的是不存在的关联边排名在k%以内的比率.当阈值k在1~100之间变化时可以得到相应的TPR和FPR.通过这种方式,可以绘制ROC曲线,从而计算AUC.按照以上步骤,我们在lncRNA-疾病异质信息网络上进行了留一交叉验证,并取得了0.682 8的AUC.相应的ROC曲线如图4所示:
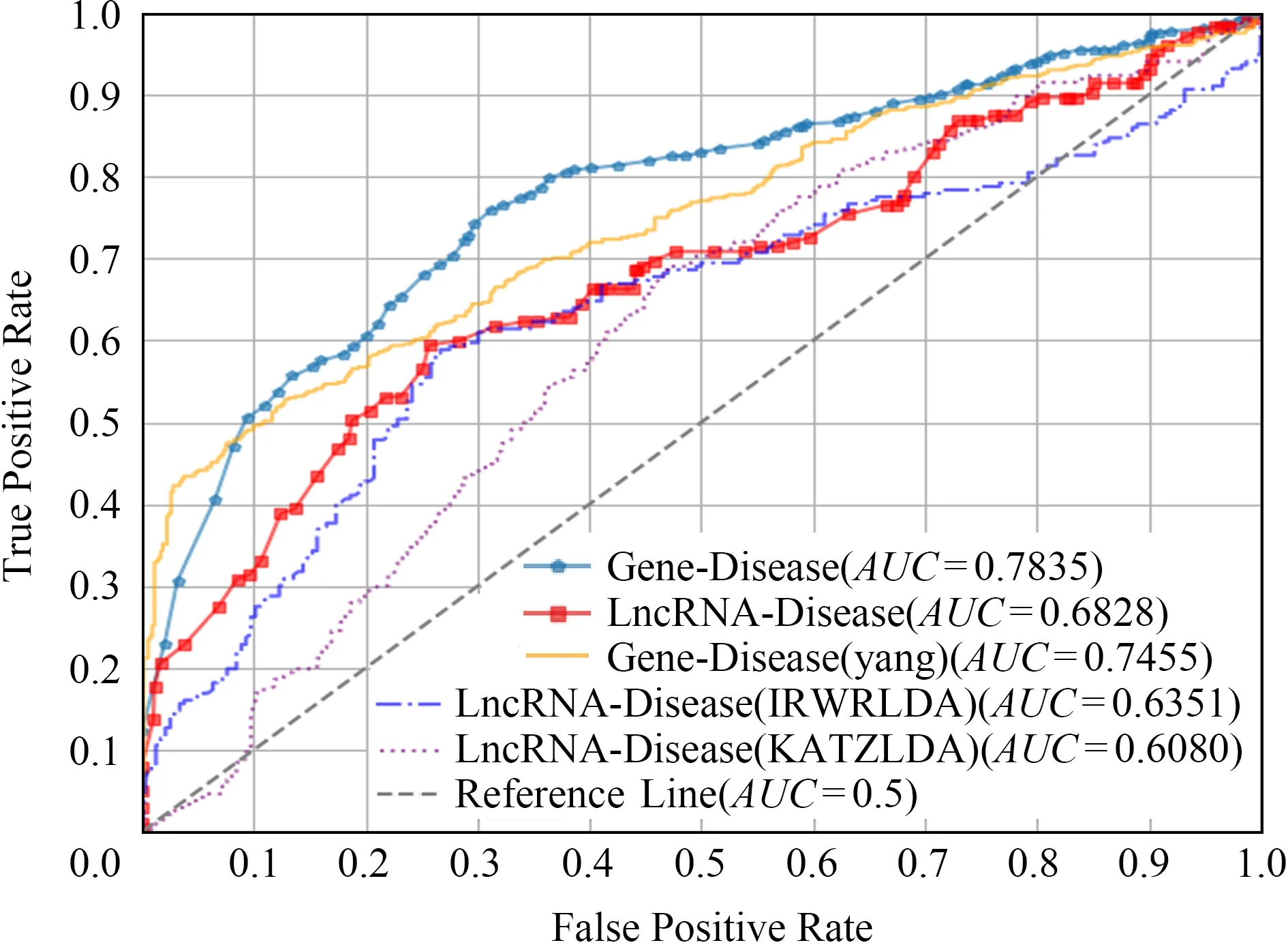
Fig. 4 Leave-one-out cross validation ROC curve图4 留一交叉验证ROC曲线图
为了提高方法的性能,我们将编码基因和疾病关联关系整合到lncRNA-疾病网络中得到基因-疾病异质信息网络.我们在基因-疾病异质信息网络上进行了留一交叉验证,负样本的构造方法与之前类似,得到的AUC值为0.783 5,如图4所示.很明显,编码基因-疾病关联关系的整合可以提高我们方法的性能,分析原因主要是通过集成编码基因-疾病关联数据增加了网络中边的数量,使网络结构变得更紧密,潜在的基因可以从其他基因和疾病中获得更多信息传播,从而可以更好地进行预测.因此,在我们做链路预测相关方面研究时,通过整合多种数据,结合更有意义的语义信息,可以有效地提升预测的准确性.
在这里我们与Yang等人[25]提出的方法在相同的数据集上进行比较,这2种方法都是基于已知的基因与疾病之间的关联,不借助其他的信息进行疾病与基因的关联预测,图4给出了本文方法与Yang等人的方法预测结果比较,本文方法优于Yang等人的方法.此外.我们又与IRWRLDA[21]和KATZLDA[22]这2种方法进行比较,这2种方法除了已知的lncRNA-疾病关联数据,还加入了lncRNA相似性和疾病相似性的数据来进行预测,本文的方法优于这2种方法,比较结果如图4所示.
2.3 案例分析
为进一步验证本文方法的可靠性和实用性,分别对卵巢癌和胃癌2种疾病做案例分析.对每一种疾病,所有未与该疾病有关联连边的基因按照其与该疾病的关联得分从大到小进行排序,排名top10的基因被认为是与该疾病潜在关联的基因.
卵巢恶性肿瘤是女性常见的恶性肿瘤之一,发病率仅次于子宫颈癌和子宫体癌.而卵巢上皮癌死亡率占各类妇科肿瘤的首位,对妇女的生命造成非常严重的威胁.表2显示了卵巢癌中排名top10的基因,包括4个lncRNA,目前这4个已有文献通过生物实验等证实确实与该疾病有关,对应的PubMed唯一标识码(PubMed unique identifier, PMID)也在表2中给出,通过PMID可以在PubMed搜索引擎中查阅对应的文献.例如:Zhou等人[27]通过研究发现MALAT-1在卵巢肿瘤中高表达,会促进卵巢癌细胞的生长和迁移,表明MALAT-1可能是卵巢癌发展的重要因素;Yang等人[28]通过实验发现UCA1在上皮性卵巢癌组织和细胞中异常上调,研究表明UCA1是上皮性卵巢癌的新预后生物标志物;Xiu等人[29]发现MEG3的表达在上皮性卵巢癌中较低,通过调节ATG3活性和诱导自噬在上皮性卵巢癌中充当肿瘤抑制剂,并可能被认为是卵巢癌的生物标志物;Zhang等人[30]研究发现在患有卵巢癌的患者中,HOTAIR显著上调.此外,HOTAIR的上调增加了卵巢癌细胞的增殖、迁移和侵袭,从而促成了卵巢癌细胞的恶性进展.

Table 2 Top10 Genes Linked to Ovarian Cancer表2 Top10与卵巢癌有关的基因
胃癌是起源于胃黏膜上皮的恶性肿瘤,在我国各种恶性肿瘤中发病率居首位,对人类的健康造成巨大威胁.表3显示了胃癌中排名top10的基因,包括5个lncRNA,其中有3个目前已有文献证实确实与该疾病有关.例如:Okugawa等人[31]通过实验发现在腹膜播散的胃癌细胞中,HOTAIR的SiRNA抑制细胞增殖、迁移和侵袭,为HOTAIR表达作为鉴定腹膜转移患者的潜在生物标志物的生物学和临床意义提供了新的证据,并且作为胃肿瘤患者的新治疗靶点;Chen等人[32]通过实验发现MALAT-1在胃癌细胞系和组织中上调;此外,MALAT-1在高转移潜能胃癌细胞系SGC7901M中的表达高于在低转移潜能胃癌细胞系SGC7901NM中的表达,结果表明MALAT-1可能部分通过调节上皮间质转化(EMT)促进胃癌细胞的迁移和侵袭;Xu等人[33]通过实验证明MEG3miR21通过调节EMT参与胃癌的肿瘤进展和转移.
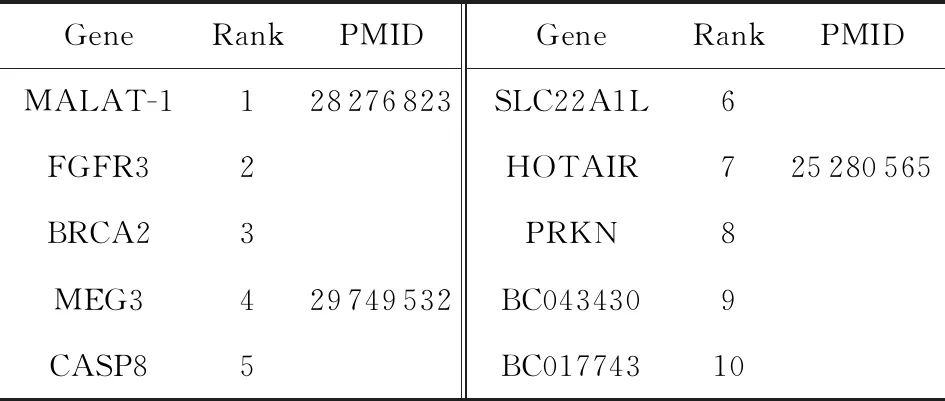
Table 3 Top10 Genes Linked to Gastric Cancer表3 Top10与胃癌有关的基因
3 结 论
长非编码 RNA在许多生物过程中具有重要的功能,这些长非编码 RNA 的变异或功能失调会导致一些复杂疾病的发生.因此,通过生物信息学方法预测潜在的长非编码 RNA-疾病关联关系,这对于致病机理的探索以及疾病诊断、治疗、预后和预防都具有重要的意义.
近年来,针对这一问题,很多研究者已提出了其他基于网络的预测方法,并且在网络模型的基础上集成基因表达数据或者基因与miRNA之间的调控关系数据,实现lncRNA与疾病关联的预测.
本文使用了一种异质信息网络中的相关性计算方法——HeteSim,用来预测lncRNA与疾病之间的关联.该方法基于路径约束,通过元路径两端节点随机游走到中间节点相遇的概率作为疾病与lncRNA之间的关联得分,发掘潜在的疾病与lncRNA关联关系.实验结果表明该计算方法有较高的预测准确性和鲁棒性,并且该方法可以很好地集成其他类型的关联数据,例如基因间的蛋白质相互作用[34]、lncRNA和编码基因的共表达、miRNA对lncRNA和编码基因的调控、疾病之间的相似性信息等.集成这些关联数据,从而对元路径进行扩展,可以使更多与lncRNA疾病相关的语义信息被用来预测,有利于预测的准确性,这也是本文工作进一步深入研究的方向.
