系列CoMnZnZ四元Heusler化合物的结构和半金属铁磁性*
2019-09-04许佳玲贾利云靳晓庆郝兴楠马丽侯登录
许佳玲 贾利云† 靳晓庆 郝兴楠 马丽 侯登录
1)(河北建筑工程学院数理系,张家口 075000)
2)(河北师范大学物理科学与信息工程学院,石家庄 050024)
1 引 言
自从1983年由de Groot及其合作者在NiMnSb中第一次发现了半金属性,半金属材料就引起了研究者的广泛关注,如果将半金属应用于磁性随机存储器等器件可以极大地改善器件的功能[1].金属铁磁体作为改进自旋电子学器件的新材料具有广泛的应用前景,其中由于Heusler合金的半金属材料具有高居里温度,在实际应用上更为便利.Heusler化合物半金属性的研究之所以成为热点还有一个原因就是其整数型的磁矩是可以预测的.现在已经证实Heusler化合物体系中的半金属材料会遵从Slater-Pauling法则[2],这个法则给出了半金属的分子自旋磁矩(Mt)和价电子数(Zt)之间的简单关系[3]:遵从Mt=Zt–18 或者Mt=Zt–24 关系.最近又发现Mn2Cu-基的Heusler化合物遵守Mt=Zt–28 的法则[4,5],而满足这个关系法则的体系较难发现,所以报道很少.
最近,一个被称为 LiMgPdSn型的新的四元Heusler化合物结构类型在实验和第一原理计算方面受到极大的关注,此类化合物有望成为新的半金属材料[6−15].随着关于四元Heusler化合物的研究与日俱增,实验和理论研究发现四元Heusler化合物 CoFeMnZ(Z=Al,Ga,Si,Ge)是具有极高居里温度的半金属铁磁体[16,17].采用第一原理计算研究发现CoRhMnZ(Z=Al,Ga,Sn,Sb,Ge,Si)和Co0.5Rh1.5MnSb也都具有半金属性,另外对于此结构的稳定性和高压相变研究也开始成为人们关注的热点[18,19].
虽然四元LiMgPdSn型的新结构类型成为了新的研究热点,但是由于符合Mt=Zt–28 法则的体系较少,这一体系的LiMgPdSn型半金属材料未受到关注.基于以上原因,本课题组[20]通过采用局域自旋密度近似(LSDA)作为交换关联泛函计算了大量四元化合物,最终发现有8种新型化合物为半金属铁磁体并且遵从总的价电子数减去28的Slater-Pauling法则.
为了完善此Heusler型半金属体系和进一步确认四元LiMgPdSn型结构的半金属性,同时为相关实验提供理论依据,本文通过第一原理方法,基于Perdew-Burke-Ernzerhof的广义梯度近似(GGA)泛函计算了 CoMnZnZ(Z=Si,Ge,Sn,Pb)系列Heusler化合物的弹性常数、体积弹性模量,进而计算了其剪切弹性模量和杨氏模量等力学参量,并根据弹性模量计算了该系列化合物的声速和德拜温度;对此系列化合物进行了电子结构和磁性的研究,并利用轨道杂化理论对三个Heusler半金属化合物的磁性起源和Slater-Pauling法则给出解释.
2 计算方法
采用基于密度泛函理论的全势能线性缀加化平面波方法(FP-LAPW)进行计算,使用的软件为WIEN2k代码包,所用的交换关联势为在合金计算中使用最为广泛的Perdew-Burke-Ernzerhof的 GGA.在具体计算中,密度泛函数理论中的截断参数Rmt×Kmax=9(这里的Rmt是最小的muffin-tin球半径,Kmax是指平面波扩展中的最大k矢量).在muffin-tin球内的球谐函数中扩展到lmax=10.四面体网格积分中在布里渊区内选取5000个k点.在自洽循环中选择能量和电荷作为收敛标准,收敛标准为默认值.
3 结果与讨论
3.1 结构和力学性质
如果用不等价原子的位置坐标描述不同元素原子所占的位置,定义占据威克夫坐标X=(0,0,0)为A位,为B位,为C位,主族元素为D位,按照Heusler化合物元素排布的价电子规则[21],价电子较多的过渡金属元素首先占据C位,然后占据A位,价电子最少的过渡金属元素首先占据B位,主族元素优先占据D位.图1给出了LiMgPdSb型Heusler化合物的晶体结构,这一类结构的空间群为F-43m(216),晶胞结构可以看成是4个面心立方子晶格套嵌而成.本文所计算的化合物按照以上占据法则填充各个不等价晶位,即CoMnZnZ(Z=Si,Ge,Sn,Pb)这种结构中 Co 元素占据A位,Mn 占据B位,Zn 占据C位,D位为不同的主族元素.首先,对于四个LiMgPdSb型Heusler化合物,在考虑自旋极化的情况下进行了体积优化,得到了能量和体积的关系曲线.使用GGA计算总能量和体积关系通过三阶的Murnaghan状态方程[22]进行拟合,进而获得能量基态的性质.表1列出了通过体积优化得到的平衡晶格常数a0、体积弹性模量B、压力导数B'和总能量E0.从表1观察到CoMnZnPb具有最小的体积弹性模量,而CoMnZnGe和CoMnZnSi的体积弹性模量较大.关于 CoMnZnZ(Z=Si,Ge,Sn,Pb)四种化合物的基本电子结构和我们之前采用LSDA计算得到的结果[20]相同,根据使用两种不同的泛函计算结果的一致性,证实我们对于 CoMnZnZ(Z=Si,Ge,Sn,Pb)四种物质的计算是可信的.
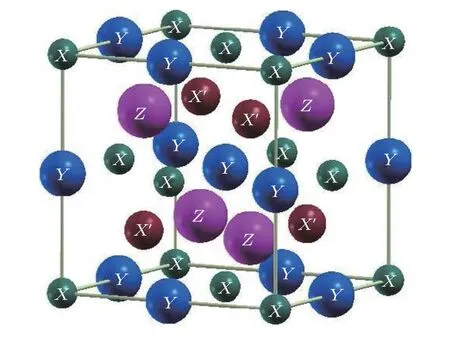
图1 LiMgPdSb 型 Heusler合金的结构示意图Fig.1.Structure of LiMgPdSn-type Heusler alloys.
通过计算弹性常数Cij探究物质的组成稳定性,这些常数在描述材料的力学性能中是不可或缺的,因为它们和各种基本固态现象密切相关,比如原子间成键、状态方程和声子谱;同时弹性性能将影响比热、热膨胀、德拜温度等热力学性能.更重要的是,弹性常数在固体力学性能的实际应用中是必不可少的:弹性常数是应变εi与应力σi之间的比例系数,σi=Cijεi,因此弹性常数可以确定作用在晶格上的外力与形变的关系,一般情况下共有21个独立的弹性常数Cij,但是由于我们研究的立方晶体的对称性使之减少为3个(C11,C12和C44),在计算弹性常数Cij时,将总能量看作体积和残余应变的函数,更详细的计算可参阅文献[23].弹性常数Cij可由 CoMnZnZ(Z=Si,Ge,Sn,Pb)单晶通过第一原理程序的弹性常数模块计算得到,然而实验制备材料常为多晶,因此对多晶而言,估算其对应的弹性模量很重要,可用以下公式推测其他的力学参数,如剪切弹性模量G、杨氏模量E、泊松比n和剪切各向异性常数A[19,24]:
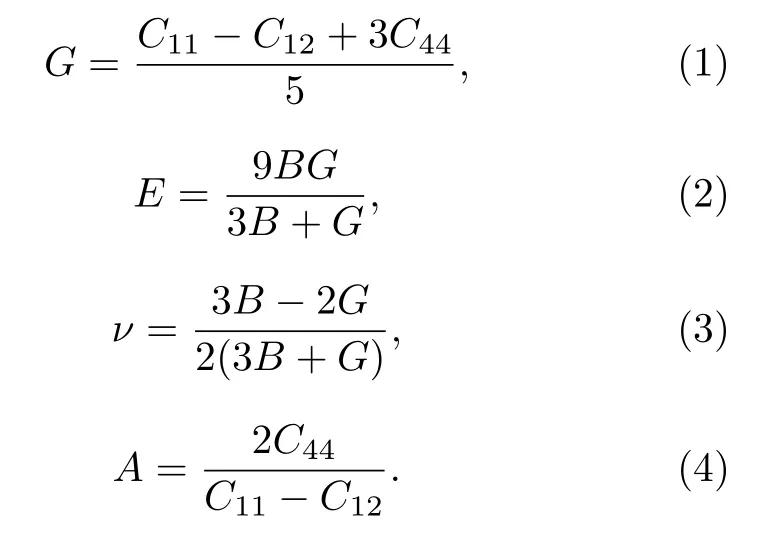
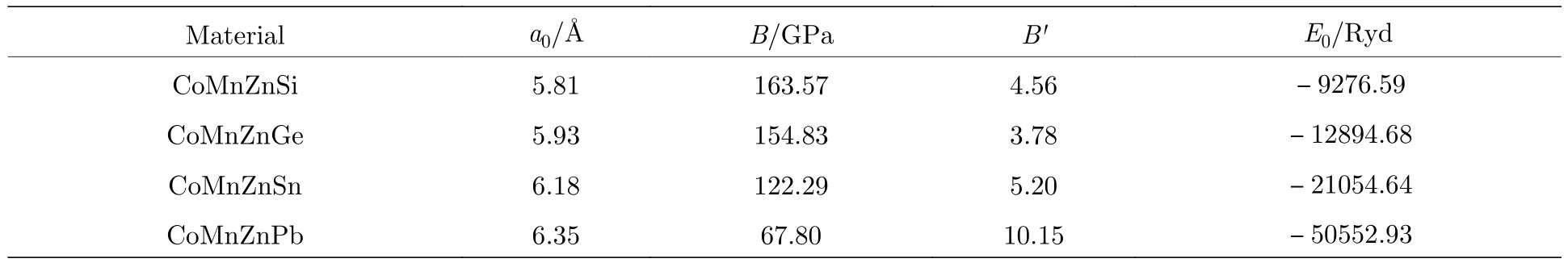
表1 CoMnZnSi,CoMnZnGe,CoMnZnSn,CoMnZnPb 四种化合物对应的平衡晶格常数 a0、体积弹性模量 B、压力导数B'和平衡能量E0Table 1. Calculated equilibrium lattice parameters a0,bulk modulus B,its pressure derivative B' and equilibrium energy E0 for CoMnZnZ(Z=Si,Ge,Sn,Pb)compounds.
计算得到的3个独立弹性常数、剪切弹性模量、杨氏模量、泊松比和剪切各向异性常数列于表2中.根据稳定性原理,我们所研究的物质在其能量最低的结构类型下力学性能更稳定,对于所有物质组成 Cauchy 应力[25,26](pc=C11−C44)恒为正.脆性和韧性等力学性能可以做如下解释:根据Pugh[27]给出的简单关系式,将材料的塑性性能与弹性模量的联系表示为B/G;对于区分脆性材料和韧性材料的临界值,Pugh认为该比例值接近1.75,如果B/G>1.75,材料表现出韧性的性能,否则材料表现出脆性性能;还有一种说法是建议将B/G=2.67作为临界指标.当然根据我们的计算,四个化合物B/G均大于5,无论采用何种判据,体系均体现出较好的韧性.另外C11的值反映沿着晶体主方向的单轴压缩,其值大于C44表明晶体承受剪切的性能弱于承受单轴压缩的性能,计算得到的体积弹性模量和杨氏模量表明,这些物质组成具有较强的不可压缩性,一般认为离子材料的典型泊松比n为0.25[28],计算中得到的物质泊松比大于0.25,表明金属键对于化合物的组成有较大的贡献.根据GGA计算得到物质的剪切各向异性因数列于表2中,对于各向同性晶体而言,这个因数A为1,小于或者大于这个数值都是由于剪切各向异性所导致,负值表明这些组成具有较低的剪切各向异性性能,故决定了在其实验合成的过程中具有较低的概率出现微裂纹和结构缺陷[29].
根据计算得到的杨氏模量E、体积弹性模量B和剪切弹性模量G,可根据平均声速vm估计德拜温度[30]:
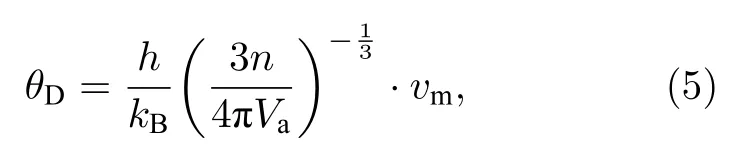
式中h为普朗克常数;kB为玻尔兹曼常数;Va是原子体积;多晶材料中平均声速由参考文献[30]给出,
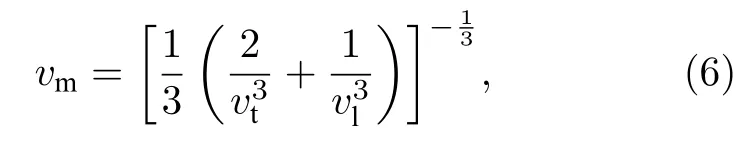
这里vl和vt分别是纵向和横向声速,通过剪切弹性模量G和体积弹性模量B由维纳方程[19]求得,
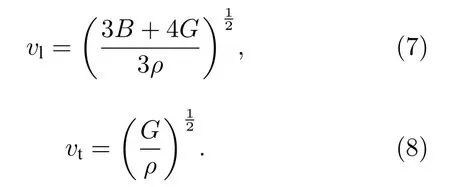
在稳定结构状态对CoMnZnSi,CoMnZnGe,CoMnZnSn和CoMnZnPb计算得到的声速和德拜温度在表3中列出,CoMnZnPb表现出最低的德拜温度,CoMnZnGe得到最高值.
表2 计算得到的各化合物的单晶弹性常数Cij、多晶剪切模量G、杨氏模量E、泊松比n和剪切各向异性常数ATable 2. Calculated single crystal elastic constants Cij and polycrystalline elastic modulus(shear modulus G,Young’s modulus E,Poisson’s ratio n)shear anisotropic factor A for compounds.

表3 计算得到的温度压力均为0状态下的纵向(vl)、横向(vt)、平均声速(vm)和德拜温度(qD)Table 3. Calculated longitudinal(vl),transverse(vt),and average(vm)sound velocity and Debye temperature(qD)for compounds.
3.2 电磁性质
利用GGA方法计算系列化合物CoMnZnZ(Z=Si,Ge,Sn,Pb)的电子结构特性,通过计算得到的自旋向上和自旋向下的总态密度(TDOS)和分波态密度(PDOS)来表示.化合物CoMnZnSi,CoMnZnGe,CoMnZnSn 和 CoMnZnPb 的 TDOS和PDOS分别如图2所示.从这些结果中,我们注意到 CoMnZnSi,CoMnZnGe,CoMnZnSn 三个样品在平衡晶格常数都显示出半金属的态密度特性;但是CoMnZnPb化合物在平衡态时自旋向下的态密度并不为零,即显示为非半金属.我们也用 LSDA做了对应的计算,虽然采用GGA优化得到的平衡晶格常数比LSDA的要大,但是自旋向下的半金属带隙量值并没有增大,化合物CoMnZnSi两种泛函得到的半金属带隙是一样的,均为0.66 eV.而CoMnZnGe化合物采用GGA得到较大的晶格常数却得到更小的半金属带隙[20].从TDOS中可以把研究的TDOS结构分成两个区域:一个区域为低于–7 eV和高于5 eV以外的范围,这个区间的态密度从PDOS分析可知是主要来自于s,p元素Si/Ge/Sn/Pb和满壳层Zn的贡献;另外一个区域是处在–7 eV和 5 eV之间的部分,表现出强烈的 3d元素 Co,Mn的杂化.这些 3d过渡金属的d态杂化不仅产生了带隙而且是产生半金属性的原因.为了理解3d元素Co,Mn的杂化以及半金属性的物理根源,在图3中给出了自旋向下部分的轨道示意图,对照图2中前三个半金属的PDOS,轨道名称之前标注的数字表示简并度.主族元素Si,Ge或Sn有1个s轨道和3个p轨道,而且能量均已经填满;Zn均有5个3d轨道而且也已经填满,其自旋磁矩几乎为零.半金属带隙主要来自于Co和Mn原子之间的相互作用,当Co和Mn由于晶体场劈裂形成d-d 杂化时,其d轨道分裂为1个二重简并eg态和1个三重简并的t2g态.正如图3中所示,Co的t2g与Mn的t2g杂化形成三重简并的t2g填充费米面以下的d杂化轨道以及费米面以上未填充的三重简并的t2g杂化轨道;eg轨道和 t2g轨道类似,形成已填充的二重简并的eg轨道和未填充的二重简并的eg杂化d轨道.因此,自旋向下的能带共包含 14个已填充态:Si,Ge或Sn 的 1×s和 3×p,Zn 原子的 2×eg和 3×t2g,Co 和 Mn 的 d-d 杂化 2×eg和 3×t2g.在自旋向上的能带中,剩余的价电子完全填充.这就解释了这些化合物遵守的Mt=Zt–28 的 Slater-Pauling法则[2,3].
为了更清楚地了解磁场属性,研究了化合物的总磁矩和局域磁矩.各个磁矩值列在表4中,是经过电子结构计算得到的总磁矩,前3个样品中的计算值和根据 Slater-Pauling 准则中的Mt=Zt–28得到的结果完全一致,通过态密度图也证明CoMnZnSi,CoMnZnGe,CoMnZnSn 三个化合物都是半金属铁磁体,而且都是在自旋向下方向出现带隙.最后一个CoMnZnPb化合物磁矩与Slater-Pauling法则预测曲线出现偏离,磁矩并非整数且没有实现100%自旋极化,表明其为非半金属.分析计算得到的各个特定元素磁矩的值可知,磁性的贡献主要来自于Co和Mn原子,尤其是Mn的作用更大,导致产生最高的磁矩值.所计算的四个化合物中贵金属Zn和主族元素Z原子的磁矩极小,几乎可以忽略不计.这与前面态密度图和轨道填充示意图分析结果一致.
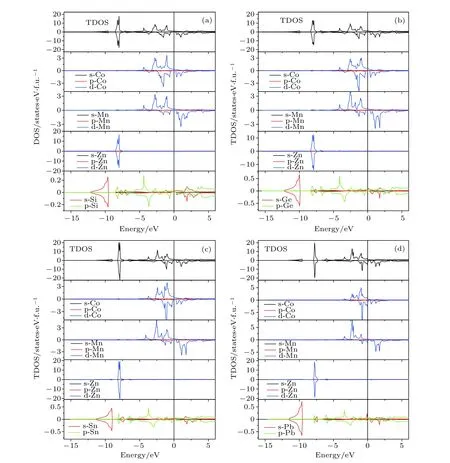
图2 四个化合物的态密度图,其中最上方的是TDOS,下方分别为各原子的PDOS(a)CoMnZnSi;(b)CoMnZnGe;(c)CoMnZnSn;(d)CoMnZnPbFig.2.Totals and Partials density of states(TDOS,PDOS)in their stable structure:(a)CoMnZnSi;(b)CoMnZnGe;(c)CoMnZn-Sn;(d)CoMnZnPb.
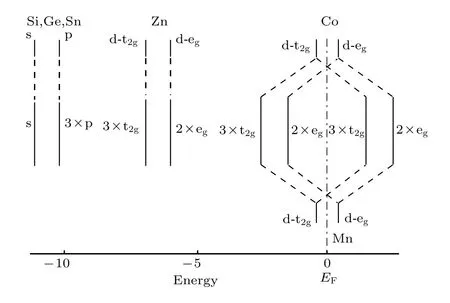
图3 自旋向下轨道填充状态示意图Fig.3.Sketch of the spin-down orbital’s occupied states.
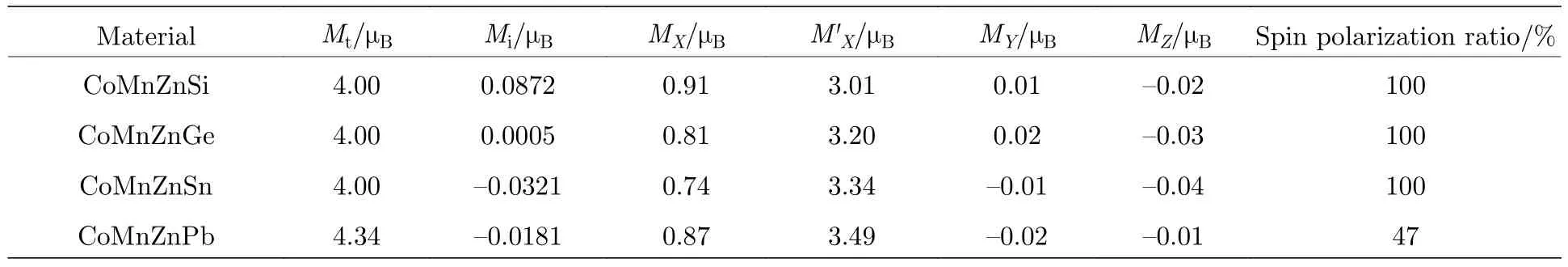
表4 每分子式总自旋磁矩 Mt、间隙区磁矩 Mi和各原子磁矩(MX,M'X,MY,MZ)、自旋极化率Table 4. Total,interstitial and local magnetic moments,calculated spin-polarization.
4 结 论
通过第一原理计算理论预测了CoMnZnZ(Z=Si,Ge,Sn,Pb)系列 Heusler化合物的力学性质、电子结构和磁性.力学性质计算结果显示化合物抗弹性变形稳定,在制备过程也不易出现微裂纹和结构缺陷;非半金属的CoMnZnPb表现出最低的德拜温度,而CoMnZnGe得到最高值.电子结构计算显示 CoMnZnZ(Z=Si,Ge,Sn)三个化合物属于半金属铁磁体,但是CoMnZnPb化合物并不显示半金属性.此外 CoMnZnZ(Z=Si,Ge,Sn)三个化合物的磁矩通过Slater-Pauling法则计算得到的量值与第一原理计算得到的完全一致,即磁矩为整数且自旋极化率为100%.通过态密度图和原子磁矩数值发现3d元素Co,Mn 对半金属化合物的磁性贡献明显,并且利用轨道杂化理论解释了磁矩为整数并符合Slater-Pauling法则的原因.综上所述,CoMnZnZ(Z=Si,Ge,Sn)三个半金属铁磁体的磁性和自旋极化率使其有望成为制作自旋电子学器件的备选材料.
