内蒙古玉米秸秆还田土壤细菌多样性特征
2019-08-30萨如拉杨恒山高聚林宋桂云李媛媛
萨如拉,杨恒山,高聚林,宋桂云,李媛媛
(1.内蒙古民族大学农学院,内蒙古饲用作物工程技术研究中心,内蒙古 通辽 028000;2.内蒙古农业大学农学院,内蒙古 呼和浩特 010018)
秸秆还田是改善农田生态环境,发展持续农业、旱作农业的重大措施;内蒙古地区每年均有大量玉米秸秆被作为柴草烧掉或在田头烧掉,进行秸秆还田规模较少;冷凉地区冬季时间长、气温低,导致秸秆腐熟不完全;温度成了冷凉地区推广秸秆还田技术的主要瓶颈。张星等[1]的研究表明,土壤微生物量碳、氮均与土壤温度显著正相关,而与土壤水分的相关性较差,玉米生育期土壤温度是影响土壤微生物量变化的主要因素之一。李晓莎[2]的研究中秸秆还田主要对0~10 cm土层微生物起作用,但匡恩俊等[3]的研究中秸秆还田下层土壤微生物量碳、氮的含量高于中、上层土壤。冀保毅等[4]的研究表明,秸秆还田能增加整个耕层土壤微生物数量;顾美英等[5]的研究中不同秸秆还田方式均能显著提高和田沙化土壤微生物活性和功能多样性,秸秆直接粉碎还田有增加土传病害的风险。于寒[6]研究表明,在玉米连作种植方式下,秸秆深埋可增强微生物代谢活性。而李涛等[7]的研究表明,秸秆还田对土壤微生物功能多样性指数没有显著性影响。目前关于秸秆还田土壤微生物效应多样性的影响还没有一致的定论,主要原因是土壤微生物特性的影响因素互同作用很复杂。因此,有必要分地区研究秸秆还田后土壤微生物特性的变化,为秸秆腐熟剂研制提供理论参考。
1 材料与方法
1.1 试验地概况
试验地设在内蒙古农业大学科技示范园区(土默特右旗萨拉齐镇)秸秆还田定位试验田,砂壤土。试验区属半干旱中温带大陆性季风气候,年均气温6~8℃,年降水总量400 mm,无霜期140 d,海拔1 015 m,年日照时数2 806 h,年活动积温为3 000~3 500℃。试验地有机质含量20.03 g/kg,碱解氮57.74 mg/kg,有效磷7.77 mg/kg,速效钾89.33 mg/kg;基施 N 40.5kg/hm2、P2O5103.5kg/hm2、K2O 45kg/hm2,拔节期一次追施N 172.5kg/hm2,生育期间灌水4次。
1.2 试验设计
2013年设玉米秸秆深翻还田一年(SF-Ⅰ)和常规旋耕秸秆不还田(CK)、2012~2013年连续两年玉米秸秆深翻还田(SF-Ⅱ)3个处理,3次重复,玉米秸秆还田方式为秋季全量(折干物质6 750kg/hm2)粉碎(秸秆长度2~3 cm)还田,还田深度30 cm。玉米品种为郑单958,种植密度7.5万株/hm2,小区面积214 m2。
播种期、苗期、拔节期、大喇叭口期、灌浆期、成熟期进行采样。本研究区耕层较浅(20 cm左右),半熟化层在耕作层以下20 cm左右,0~20、20~40 cm土层采用5点取样法,按对角线取样;同一处理同一生育期土样分别混合均匀,3个处理6个时期2个土层样品共36份,按四分法取100 g土壤样品装入已灭菌的自封袋中,将新鲜土样分别过2 mm筛用于土壤总DNA提取,风干土壤用于测定土壤有机质和土壤纤维素酶活性。
1.3 测定项目与方法
土壤细菌群落16S rDNA-PCR-DGGE分析采用文献[8]方法进行。
土壤有机质采用重铬酸钾容量法-外加热法[9]测定。
土壤纤维素酶活性用3、5-二硝基水杨酸比色法[10]测定。
1.4 土壤细菌16S rDNA文库构建
DGGE聚类分析筛选出典型样品,其总DNA为模板采用细菌16S rDNA通用引物(63F/1492R)对土壤细菌16S rDNA进行PCR扩增,扩增体系为10×PCR Buffer 2μL、dNTP 1.6μL、27F 0.2μL、1492R 0.2μL、Taq酶0.1μL、土壤DNA稀释50倍1μL、无菌水14.9μL;PCR扩增反应程序为94℃变性3 min、94℃变性30 s、62℃退火40 s、72℃延伸50 s、35次循环,72℃延伸5 min。
将扩增片段与pEASY-T1载体(0.5μL载体,4.5μL DNA) 连接后转入Ecoli.DH5a中,涂LBAmp(100μg/mL)平板上37℃培养16 h后,随机挑取克隆子,通过菌落PCR检测阳性克隆子,即应用pEASY-T1载体通用引物M13(+)、M13(-)重新扩增插入的片段,进行克隆文库的菌落PCR,菌落 PCR反应体系为10×PCR Buffer 2μL、dNTPs 1.6μL、M13+(20 μm)0.2μL、M13-(20μm)0.2μL、Easy Taq DNA聚合酶0.1μL、模版1μL、无菌水14.9μL;菌落PCR反应程序为94℃变性3 min、94℃变性45 s、55℃退火30 s、72℃延伸90 s、30次循环,72℃延伸5 min。
分别建立3个典型样品细菌16S rDNA 3个克隆文库。随机挑取阳性克隆,阳性克隆子上海生物工程有限公司测序。测序后先将核酸序列中的载体序列进行手动删除,在NCBI GenBank数据库中Blastn程序进行相似性比对,确定最相似参比序列。将每个文库已测的16S rDNA序列和从GenBank数据库中比对的最相似参比序列,用ClustalX2软件进行多重比对比齐,截齐后的序列用MOTHUR软件进行分析,以cutoff=0.05为分界阈值划分OTU。
运用Quantity One软件(Bio-rad,US)对DGGE指纹图谱进行分析。UPGMA(Unweighted pairgroup method using arithmetic averages)法进行相似性聚类分析,同时输出各条带位置及光密度值。将各条带光密度值进行标准化处理后,计算多样性指数。
1.5 数据处理
数据采用Excel 2003及DPS 7.05软件显著性差异检验。
2 结果与分析
2.1 土壤有机质
从表1可知,随着玉米生育期的推进,0~20 cm土层,CK和SF-Ⅰ土壤有机质含量呈先增加后下降趋势,在大喇叭口期最高,成熟期最低。而SF-Ⅱ有机质含量播种期最高,成熟期最低;同一生育期,土壤有机质含量均高于CK,秸秆深翻还田能提高土壤有机质;玉米生育期内,2个秸秆还田处理有机质含量存在差异;与CK相比,SF-Ⅰ提高土壤有机质幅度在大喇叭口期(25.02%)最大;SF-Ⅱ提高土壤有机质幅度在播种期(59.96%)最大;播种期和大喇叭口期SF-Ⅰ与SF-Ⅱ有机质含量有极显著差异,其他生育期无显著差异。
从表1看出,随着土层的下移土壤有机质含量呈逐渐降低的趋势;20~40 cm土层中,播种期秸秆深翻还田处理与CK土壤有机质含量差异极显著,其他生育期无显著差异。
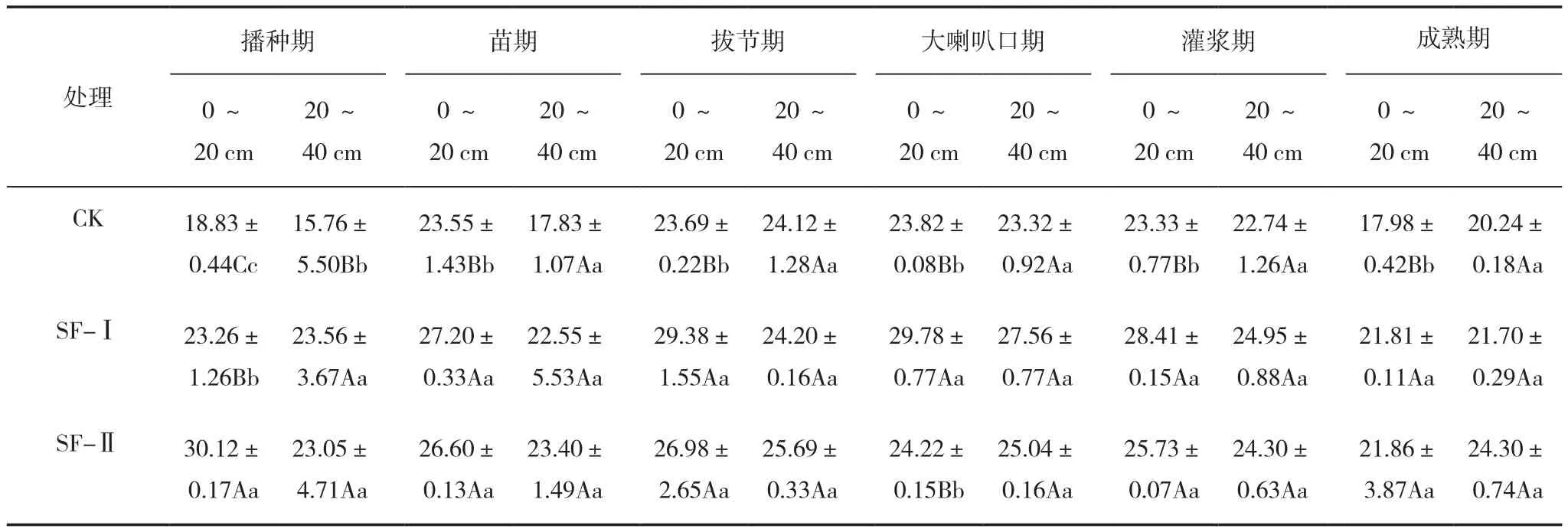
表1 玉米生育期内0~40 cm 土层平均有机质含量变化 (g/kg)
2.2 土壤纤维素酶活性分析
由表2可知,玉米生育期,0~20 cm土层纤维素酶活性表现为SF-Ⅱ>SF-Ⅰ>CK;播种期至大喇叭口期,SF-Ⅱ显著提高土壤纤维素酶活性,SF-Ⅱ比CK分别高46.51%、39.66%、82.69%、37.50%。拔节期至大喇叭口期,SF-Ⅰ和SF-Ⅱ土壤纤维素酶活性差异极显著,SF-Ⅱ土壤纤维素酶活性比SF-Ⅰ分别高出45.26%、14.29%;其余生育期无显著差异。

表2 玉米生育期内0~40 cm 土层纤维素酶活性变化 [C6H6O6 mg/(g·72 h)]
从表2看出,随着土层的下移土壤纤维素酶活性呈逐渐降低的趋势;20~40 cm土层中,拔节期SF-Ⅱ与CK土壤纤维素酶活性差异极显著,但SF-Ⅰ与CK间无显著差异;其他生育期3个处理间无显著差异。
2.3 土壤细菌群落16S rDNA-PCR-DGGE分析
由图1、表3可知,36个样品土壤细菌16S rDNA DGGE条带数、条带位置和粗度存在一定差异;玉米关键生育期0~20 cm土层条带数均多于20~40 cm。播种期0~20 cm土层中3个处理共有条带为1、2、3、6、15号,13号条带为SF-Ⅰ和SF-Ⅱ共有,2、6、10号条带在SF-Ⅱ中较粗,说明SF-Ⅱ优势菌属为此3个条带代表的菌属;20~40 cm土层中SF-Ⅰ和SF-Ⅱ与CK相比,4、14号条带变弱,甚至消失,但多了15号条带,SF-Ⅱ中出现3、13号条带,说明秸秆还田后微生物多样性增加了。玉米关键生育期0~40 cm土层DGGE条带数总体为 SF-Ⅱ>SF-Ⅰ >CK。

图1 玉米关键生育期土壤细菌16S rDNA DGGE图谱
由图2、表3可知,36个样品可归为三大类群,8~10号和19~25号共10个样品聚为一大簇,相似性指数为54%,样品均为表层(0~20 cm) 样 品;18、28、30、32、33、35~ 37号 共8个样品聚为一簇,相似性指数为52%,样品均为深层(20~40 cm)气温较低时期的样品;7、11、14、29和31共5个聚到一起,再与2~ 6、12、13、15~17、34等样品共16个聚为另一大簇,相似性指数分别为58%、56%,样品均为深层(20~40 cm)高温时期的样品;26和27号样品不能与其余样品聚到一起。从聚类分析可知,玉米秸秆深翻还田表层土(0~20 cm)与深层土(20~40 cm)样品在聚类位置上产生较大的差异;在苗期和成熟期,同土层各处理样品聚类位置相近,拔节期至灌浆期聚类位置相对较远;在播种期CK与SF-Ⅱ聚类位置远,分别在两大类里;拔节期各处理表层土壤样品(17、18、19)聚类位置远,分别在3个不同大类里;大喇叭口期各处理样品(14、15、16)聚类位置也较远,分到3个小类里。表明秸秆还田后拔节期至灌浆期土壤细菌群落发生较大的变化;在拔节期变化最大。
表3 DGGE凝胶中土壤样品编号

图2 土壤细菌16S rDNA DGGE聚类图
2.4 典型样品细菌16S rDNA文库构建
以上分析可知,拔节期各处理表层土壤纤维素酶活性较高,样品涵盖的信息多,土壤可作为典型样品。构建典型样品土壤细菌16S rDNA文库。由表4可知,150条序列共划分为59个OTU,CK样品中所建立的文库分布在20个OTU中;分别属于变形菌门(Proteobacterium)的小梨形菌属(Pirellula)、水单胞菌属(Aquimonas)、德沃斯氏菌属(Devosia)、副球菌属(Paracoccus),β-变形菌纲(Beta proteobacterium),放线菌门(Actinobacteria)的节杆菌属(Arthrobacter)。CK样品中出现了有机物降解作用的节杆菌属菌群,还出现了反硝化作用的副球菌属,厌氧氨氧化作用的小梨形菌属。SF-Ⅰ样品中所建立文库分布在25个OTU中;分别属于a-变形菌纲(Alpha proteobacterium)、酸杆菌门(Acidobacteria)、疣微菌门(Verrucomicrobia)的不可培养的微生物和地杆菌(Geobacter)、磁螺菌属(Magnetospirillum)、纤维弧菌属(Cellvibrio)、浮霉状菌属(Planctomyces)、根瘤菌属(Mesorhizobium)。SF-Ⅱ样品中所建立的文库分布在27个OTU中,分别属于β-变形菌纲(Beta proteobacterium)、γ-变形菌纲(Gamma proteobacterium)、酸杆菌门(Acidobacteria)、疣微菌门(Verrucomicrobia)、芽单胞菌门(Gemmatimonadetes)、拟杆菌门(Bacteroidetes)的不可培养微生物和可培养的变形菌门的生丝微菌属(Hyphomicrobium)、鞘氨醇单胞菌(Sphingomonas)、根瘤菌属(Sinorhizobium meliloti)、黄杆菌(Luteolibacter)、黄单胞菌属(Xanthomonas-like)、纤维弧菌属(Cellvibrio)、芽孢杆菌属(Bacillus)、短芽孢杆菌属(Brevibacillus)。SF-Ⅰ和SF-Ⅱ样品OTU比对序列中出现了纤维素分解菌和有机污染物降解菌,有芽单胞菌(Gemmatimonadetes)、短芽孢杆菌属(Brevibacillus)、芽孢杆菌属(Bacillus)、浮霉状菌属(Planctomyces)、纤维弧菌属(Cellvibrio)、鞘氨醇单胞菌(Sphingomonas)、地杆菌(Geobacter)、生丝微菌属(Hyphomicrobium)。

表4 典型土壤样品细菌16S rDNA比对序列特征
2.5 土壤细菌16S r DNA多样性指数
群落中生物种类增多代表了群落的复杂程度增高,即H*-Shannon-wiener指数值愈大,群落所含的信息量愈大。chaol指数是用来反映物种丰富度的指标,表5可知,H*-Shannon-wiener指数和Chao1值均为 SF-Ⅱ>SF-Ⅰ>CK。

表5 土壤细菌16S rDNA基因多样性指数
3 讨论
秸秆还田后土壤肥力明显提高[11],这对高产和保持土壤供养分能力具有重要意义。本试验中秸秆深翻还田提高表层土壤有机质,主要是由于秸秆施入土壤后,可以有效减少径流,使得表层养分含量得以保留,有机质含量也增加;而CK有机质含量偏低是由于玉米收获后未留下残茬,返还到土壤中的有机物较少。连续两年深翻秸秆还田纤维素酶活性最高峰值出现时期提前到拔节期;秸秆还田提高全生育期土壤纤维素酶活性;这与王静等[12]、景音娟等[13]的研究结果相同。前人应用DGGE检测群落多样性、丰度和均匀度,认为指纹分析技术中DGGE最为敏感[14-19]。本研究通过16S rDNA文库构建可知,秸秆还田提高土壤中纤维素降解细菌数量及多样性,玉米关键生育期连续两年深翻秸秆还田多样性指数最高,因为不同种植栽培方式对细菌物种优势度有影响[20-21];土地利用类型的变化造成微生物群落结构变化[22];土壤有机质是土壤微生物群落变化的重要因素[23];影响细菌群落的主要环境因素是有机质>速效氮>速效钾>速效磷[24];土壤微生物与土壤理化性质相关[25];深翻秸秆还田显著提高表层土壤有机质含量,秸秆还田后秸秆释放出氮素,氮添加有助于提高大多数功能群丰度[26]和增大土壤细菌群落多样性[27],从而增加了土壤纤维素降解菌丰度;土壤中氮源的多少和种类将影响到土壤微生物的群落结构[28]。秸秆还田循环利用显著增加了土壤微生物数量和生物量[29];糖类和聚合物类是区分土壤微生物群落功能多样性的主要碳源类型[30],可推测,秸秆还田后可能改变了土壤中糖类和聚合物类。
4 结论
秸秆深翻还田后土壤纤维素酶活性明显增强,显著提高土壤有机质含量;低温时期和高温时期土壤细菌群落差异显著,秸秆深翻还田提高了土壤细菌多样性。
