N型甲烷水合物结构和电子性质的密度泛函理论计算*
2019-08-29罗强杨恒郭平赵建飞
罗强 杨恒 郭平‡ 赵建飞
1)(西南石油大学理学院,成都 610500)
2)(西南石油大学,油气藏地质及开发工程国家重点实验室,成都 610500)
1 引 言
天然气水合物作为一种清洁、高效的非常规能源,近年来得到了许多国家的高度重视和大力发展[1-3].目前,对水合物的研究主要集中在温度[4]、压强[5]和表面活性剂[6]及CO2对水合物的置换等方面的实验研究[7-10],并通过实验方法对3种类型的水合物结构给出X-ray[11],Raman[12]谱.尽管实验研究取得了长足进展,但理论研究相对薄弱[13],在理论上,人们采用了分子动力学(MD),蒙特卡罗(MC)等方法,模拟了温度压强对水合物结构稳定性、能量和热力学性质的影响[14-21],但难以给出其电子性质.为了深入研究水合物的微观机理.人们采用密度泛函理论(DFT)对其做了研究,但主要针对结构相对简单的I型水合物(512612)水合物的结构和性质进行了研究,对II型及H型水笼子研究相对较少[22-30].
水合物是类冰状的结晶物质,在一定条件下可以稳定存在,结构受客体分子和温压的影响,所以很多高压低温的星体,存在新结构水合物的可能,探索水合物结构可以有助于认识外星球的组成.其次水合物有储存气体的能力,可以储运天然气和埋存CO2,研究新结构降低水合物形成能也是一个新的研究方向.Huang等[31]采用MC方法预测了III笼型冰相.曹潇潇等[32,33]研究了大客体烷烃分子与水笼子之间的稳定性,也做出了新的预测.
但我们发现给出的水合物结构存在五重对称轴,单个水笼子难以简单地在空间存在周期性排列,会导致运算量大、计算成本高.所以理论上寻找结构简单且具有高对称的水合物新相具有重要意义.
本研究采用密度泛函理论[34,35]第一性原理方法,构建出新型结构的甲烷水合物,并对其结构和电子性质等进行研究.通过得到结构参数、形成能、XRD图、态密度和能带,为研究天然气水合物的赋存、合成和储运等提供理论参数依据.
2 计算模型与方法
2.1 模 型
模型是受到固体物理中面心立方晶体的第一布里渊区(具有周期性、高对称性的截角八面体)启发.先做出一个截角八面体,将水分子中的氧原子置于其顶角上,甲烷分子中的碳原子放在中心,以此作为骨架,此时对称群为IMM,如图1(a)所示.采用自动加氢方式填加氢原子,由8个在平行于{111}面上(位于过体对角线1/4处)的正六边形和6个在{100}面上的正方形,构成了具有截角八面体结构的水笼子(4668),作为本研究的基本模型.通过调整氢原子的位置,再经过计算机优化后得到的结构如图1(b)所示,为了便于描述,称其为N型甲烷水合物,晶格参数为7.700 Å,密度0.903 g/cm3,大于 I型的 0.813 g/cm3、II型的0.804 g/cm3和H型的0.768 g/cm3[36].
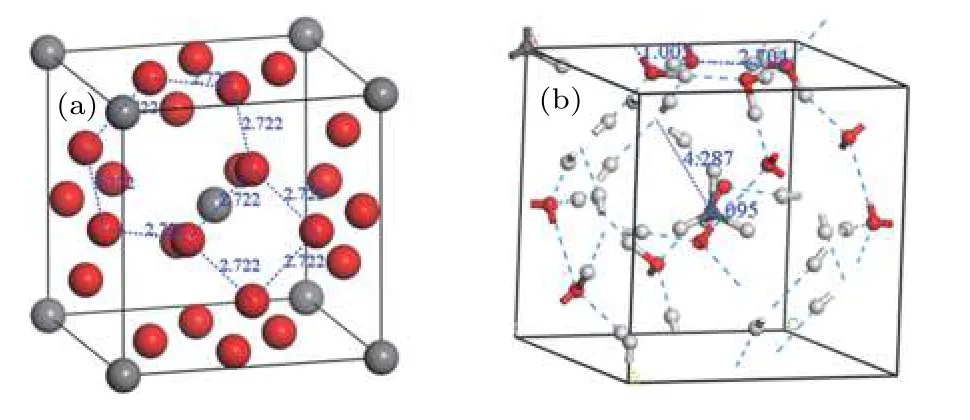
图1 模型示意图(a)N型甲烷水合物骨架模型;(b)优化后的结构Fig.1.Model diagram:(a)N-methane hydrate skeleton model;(b)optimized structure of N-methane hydrate.
2.2 计算方法及参数设置
计算采用软件是Materials Studio 2016的CASTEP[37,38]模块,它是对固体材料进行理论模拟研究的量子力学程序,由英国剑桥大学凝聚态理论研究组开发,其可靠性已经为大量实际计算所验证[39,40].本文使用CASTEP计算时,采用PBE形式的广义梯度近似(generalized gradient approximation,GGA)[41]泛函处理交换关联势进行计算,并考虑了Grimme色散修正.
经收敛性测试及计算,平面波截断能取为600 eV,K点[42]网格尺寸采用3×3×3,选择超软赝势处理电子离子间相互作用,单原子能量收敛精度为5.0×10—5eV/atom,作用于原子最大力收敛精度是0.01e V/Å,最大压力收敛精度是0.2 GPa,最大位移收敛精度是0.005 Å,能量收敛算法采用BFGS[43-46]优化算法,SCF自洽精度设为单原子能量收敛至 5.0×10—7eV.
3 计算结果及分析
3.1 优化后结构参数
由于N型甲烷水合物是一个新结构,难以与前人的研究结果进行对比,但是其结构与I型甲烷水合物较为相似,所以我们和I型甲烷水合物结构[47]进行对比研究; 另一方面对于交换关联泛函的处理,采用GGA进行计算,并采用了Grimme色散修正,优化前后晶格参数如表1所列.
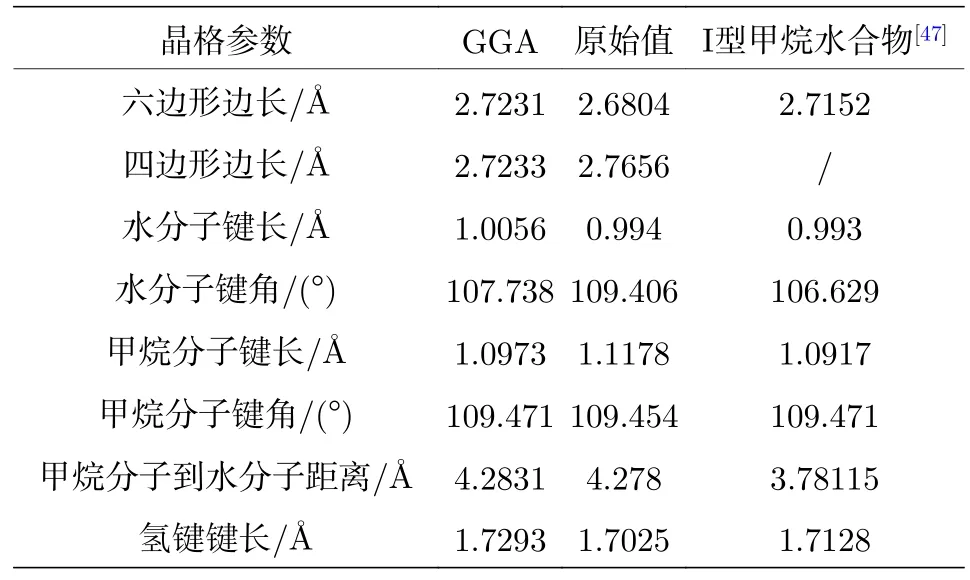
表1 平均晶格参数Table 1. Average lattice parameters.
N型甲烷水合物水笼子是由6个四边形和8个六边形构成,从表1中可以看出,优化前的六边形平均键长为2.6804 Å,优化后的六边形平均边长分别为2.7231 Å,与I型甲烷水合物水笼子中的六边形平均边长2.7152 Å较为接近; 优化前四边形平均边长为2.7656 Å,优化后为2.7233 Å; 水分子的平均键长在优化前为0.994 Å,优化后为1.0056 Å,优化前后水分子键长几乎没有发生变化,与已知I型甲烷水合物计算的0.993 Å水分子平均键长亦很接近.水分子平均键角优化前是109.406°,优化后的键角变小,为 107.738°,与 I型甲烷水合物计算的106.629°接近.
甲烷分子平均键长优化前为1.1178 Å,优化后为 1.0973 Å,比表1中的 I型甲烷水合物的1.0917 Å大,这是N型甲烷水合物水笼子比I型甲烷水合物水笼子大,水笼子与甲烷相互作用较弱导致的.甲烷分子的平均键角优化前为109.454°,优化后的结构与I型甲烷水合物值相同,说明优化并不影响甲烷分子上氢原子的位置; 甲烷分子到水分子平均距离优化前为4.278 Å,优化后为4.2831 Å,比表1中的I型甲烷水合物的值3.7812 Å要大,说明N型甲烷水合物水笼子空间更大,能容纳更大体积的气体分子.
氢键是水合物稳定性的关键因素,优化前的氢键平均键长为1.7025 Å,优化后为1.7293 Å,而表1中I型甲烷水合物参考值是1.7128 Å,I型甲烷水合物平均氢键键长较长,这可能是由于N型甲烷水合物笼子更大,甲烷分子到水分子平均距离大,甲烷影响作用弱导致.
从结构分析中可以得出,N型甲烷水合物水分子的键长键角比初始结构变化较小,这是由于水分子是极性分子,在形成水笼子的过程中出现极化,形成氢键所导致的; N型甲烷水合物中六边形平均边长,水分子平均键长、键角,氢键平均键长都比I型甲烷水合物的参数要大一些,而甲烷平均键角没有发生变化,甲烷平均键长增加,这是由于N型甲烷水合物笼子较I型甲烷水合物的大,甲烷分子与水笼子相互作用较弱,由范德瓦耳斯(van der Waals)力结合所导致的.
3.2 X射线衍射(XRD)
为了进一步了解N型甲烷水合物结构,从理论上做出了其X射线衍射图(XRD),如图2所示.由于N型甲烷水合物是从理论上提出的全新甲烷水合物结构,实验数据相对缺乏,由于与I型甲烷水合物结构比较接近,我们参考对比丁家祥等[48]I型甲烷水合物实验数据.
从图2可以看出,N型甲烷水合物在2θ值为16.5°,23.0°,28.5°,33.0°,37.0°,41.0°,44.0°和47.5°附近出现了 8个峰值,与 I型甲烷水合物XRD实验衍射图峰值中的2θ值符合较好,但比实验中的峰值个数少.这是因为实际水合物中可能存在杂质,丁家祥等[48]做的实验温度是 —90 ℃,而我们采用的是理想晶格,所选的温度趋于绝对零度,且结构本身与I型甲烷水合物结构又有所不同,这些均导致了峰值个数会出现差异.

图2 N型甲烷水合物结构X射线衍射图Fig.2.X-ray diffraction of N-methane hydrate structure.
3.3 形成能
人们认为水合物中客体分子与水笼子相互作用,是由范德瓦耳斯力作用导致的,为了考察N型甲烷水合物形成的难易程度,采用GGA进行优化计算,同时考虑Grimme色散修正,计算公式如下[45]:
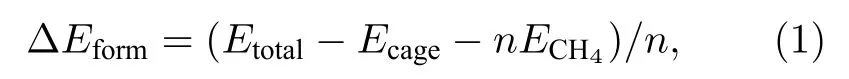
式中 ΔEform为N型甲烷水合物的形成能,Etotal为N型甲烷水合物的总能量,Ecage为N型甲烷水合物水笼子的总能量,ECH4为甲烷分子的能量,n为晶格中甲烷分子个数.根据(1)式计算出GGA近似情况下的形成能,列入表2中 ΔEform行.

表2 GGA近似下的形成能Table 2. Formation energy of GGA approximation.
表2中可看出,优化后形成能为 —0.247 eV,较I型甲烷水合物的形成能 —0.581 eV绝对值更小,表明N型甲烷水合物易形成,但稳定性比I型甲烷水合物差; 同时也反映了甲烷分子形成N型甲烷水合物的作用非常微弱,它们之间靠范德瓦耳斯力的结合形成.
3.4 态密度及分波态密度
为更好地理解N型甲烷水合物形成的机理,对其电子性质作了进一步研究,分析态密度(DOS)及分波态密度(PDOS)是研究电子性质的主要手段之一.具体针对单个甲烷分子及水笼子中的甲烷分子、水笼子与N型甲烷水合物的水笼子的态密度及分波态密度进行对比分析,其中能量为零处为费米能级,态密度的展宽宽度为0.2 eV.
3.4.1 甲烷分子态密度及分波态密度
图3为单个甲烷分子及水笼子中的甲烷分子的态密度及分波态密度,从图3可以看出,水笼子中甲烷分子的态密度较处于自由状态的单个甲烷分子,其能量分布整体向左平移; 从s态和p态电子分波态密度来看,在费米能级附近及以下有类似的变化趋势; 在能量约为5—17 eV区间,出现了明显的sp轨道杂化,且总体变化趋势朝能量更低的方向移动,这也表明了水笼子中甲烷分子能量更低,在接近绝对零度时,易形成甲烷水合物.
3.4.2 水笼子态密度及分波态密度
为了进一步理解甲烷与N型水笼子的相互作用,采用GGA近似下的态密度及分波态密度对水笼子做了分析,如图4所示.N型水笼子与空笼子的态密度图形大部分重合,主要在能量5—17 eV区间附近态密度发生了较微弱的变化,结合分波态密度来看,s态电子变化可以忽略,主要是氧原子的p态电子引起的微弱变化,这也反映出甲烷分子和水笼子的作用非常微弱,它们之间靠范德瓦耳斯力结合.
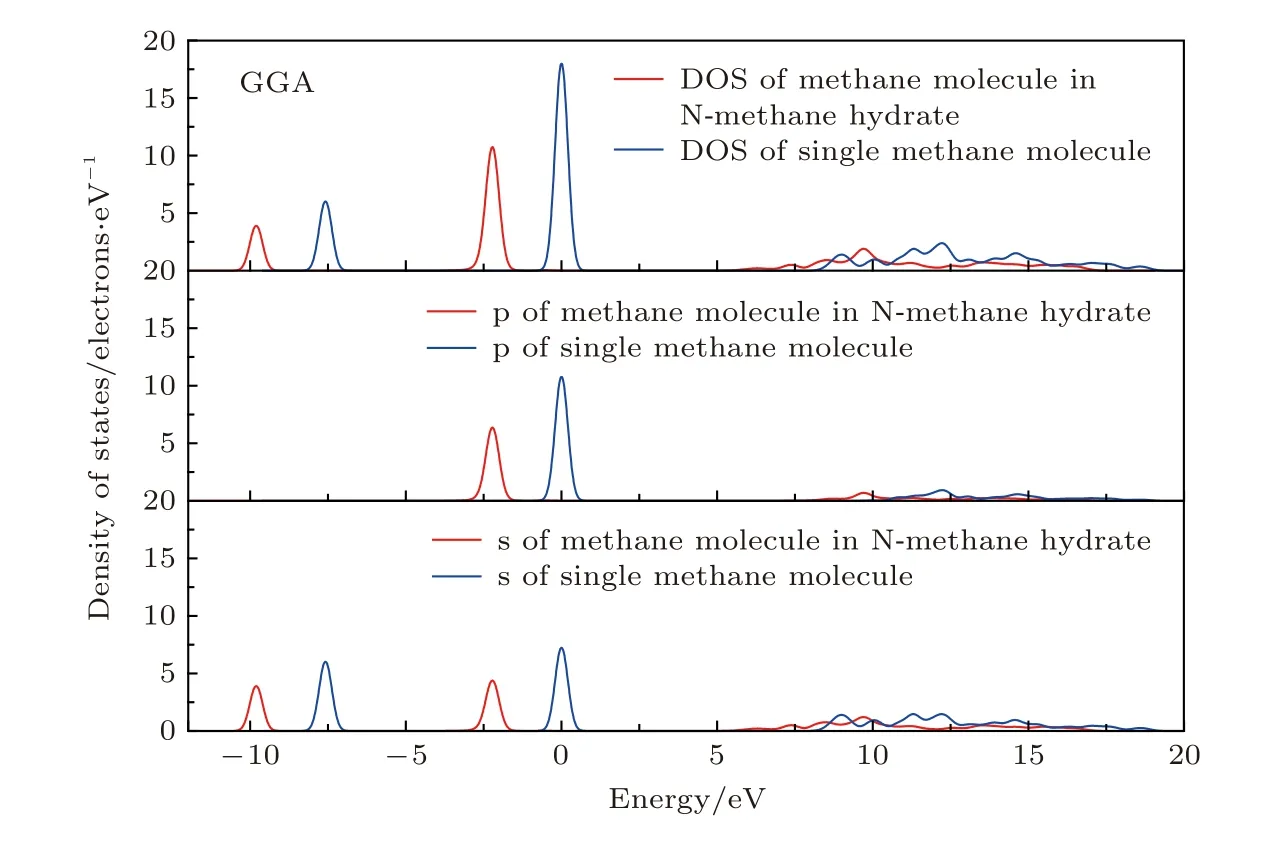
图3 甲烷分子态密度和分波态密度Fig.3.Density of states and partial density of states in methane molecules.
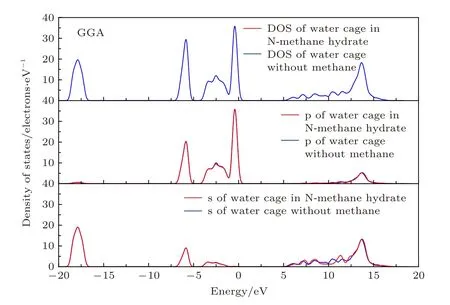
图4 水笼子的态密度及分波态密度Fig.4.Density of state and partial density of state in water cage.
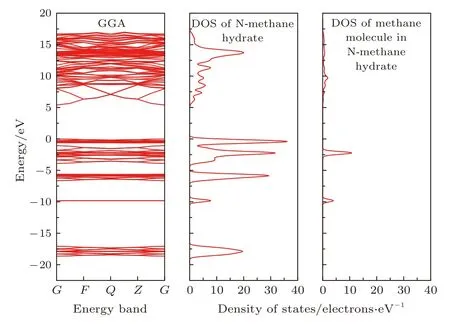
图5 能带结构Fig.5.Energy band structures.
3.5 能 带
能带结构是反映物质电子性质的另一重要概念,通过理论计算,给出了GGA近似下的能带结构,以及与之对应的N型甲烷水合物和水笼子中甲烷态密度,如图5所示.
费米能级(能量为零)以上的是导带,以下为价带,能隙(禁带宽度)为导带低与价带顶的能量之差.N型甲烷水合物的能隙为5.397 eV,价带顶与导带底位于G点处,可看为绝缘体; 结合N型甲烷水合物和水笼子中的甲烷态密度看,价带顶与导带底附近的能态密度由水笼子贡献,再结合图4中水笼子的分波态密度来看,组成水笼子中的水分子,其氧原子的p态电子对禁带宽度起了决定作用,而甲烷分子的态密度偏离带低及带顶附近,混合在水笼子的能带中,并未进入禁带,导致能隙发生改变,从这个角度来看,也体现了甲烷与水笼子的微弱作用,在宏观层面表现出是范德瓦耳斯作用.
4 结 论
基于密度泛函理论,对首次提出的N型甲烷水合物,运用MS软件中CASTEP模块研究了其微观机理.考虑Grimme色散修正,采用GGA进行对比计算,分析其结构、XRD、形成能、电子态密度和能带结构等,得到了如下结论.
1)N型甲烷水合物结构为截角八面体型的水笼子(4668),对称群为IMM,为目前已知的对称性最高的水合物水笼子,晶格参数为7.700 Å,密度为0.903 g/cm3;
2)N型甲烷水合物中六边形平均边长,水分子平均键长、键角,氢键平均键长都比I型甲烷水合物的参数要大,这是由N型甲烷水合物水笼子比I型甲烷水合物水笼子大所导致; 而甲烷平均键角没有发生变化,甲烷平均键长增加,甲烷分子与水笼子相互作用力为范德瓦耳斯力; 结合XRD来看,N型甲烷水合物与I型甲烷水合物结构较为接近;
3)N型甲烷水合物形成能、电子态密度及分波态密度均表明了甲烷与N型水笼子相互作用微弱,靠分子力作用;
4)N型甲烷水合物为绝缘体材料,能隙大于5 eV.
