胃肠道微生物种群与人类消化系统疾病相关性研究进展
2019-08-21刘德华孙宝林
刘德华, 孙宝林
(中国科学技术大学 生命科学学院, 合肥 230026)
胃肠(GI)道具有250~400 m2,是人体和环境因子及抗原的最大接触面之一[1]。在人的一生中,除了大约有60 t食物外,还有大量来自环境的微生物通过胃肠道,这些微生物对胃肠道的完整性产生了重要影响。细菌、古生菌和真菌在消化道的集落统称为肠道微生物群,它们与宿主共同进化了数千年,形成了复杂而互利的关系。据估计,人体消化道承载的微生物数量高达1014个,近乎人类细胞的10倍[2]。通过16S rRNA基因测序方法检测肠道微生物组,发现肠道微生物基因数达500万,是人类基因组的150多倍[2]。因此,通常将宿主及寄生于其上的大量微生物称为超级有机体[3]。
胃肠道作为具备代谢、免疫和内分泌功能的器官,可以与机体的其他器官相互作用、相互影响。微生物群可以增强肠道完整性、塑造小肠上皮、获取能量、抵御病原体和调节宿主免疫等。然而,这些功能可能由于微生物组成的改变而被破坏,这种肠道微生物群的改变被称为微生态失调。随着分析这个复杂生态系统的方法越来越成熟,微生物群对胃及肠道疾病的影响及作用也越来越明确。肠道微生物与消化系统疾病具有一定相关性,通过对其组成、功能以及致病机制的研究,希望有助于疾病的防治和新治疗方法的开发。本文概述了我们目前对人体胃肠道微生物群的发展和组成的认识,以及它与人体消化系统疾病的关系。
1 胃肠道微生物的研究方法
10年前,人们对胃肠道微生物群的了解大都来源于实验室大规模培养。现在,随着非培养、高通量以及低成本的测序方法的出现,对胃肠道微生物群检测能力有了很大提高。目前针对细菌16S rRNA基因的靶向性测序是检测肠道菌群的流行方法,16S rRNA基因存在于所有的细菌和古生菌中,并且包含9个高度可变的区域(V1-V9),可以依此对物种进行区分。近年来,16S rRNA基因测序已可以深入地分析该基因的短亚区,但是使用较短的读取长度(100~250 bp)可能会引入错误[4]。而宏基因组测序以环境样品中的微生物群体基因组为研究对象进行测序分析,具有较高的分辨率和灵敏度,可以更可靠地检测分析微生物群的组成和多样性。宏转录组可以直接获得环境中微生物转录组信息,对微生物群的差异表达基因和差异功能进行分析[5]。利用这些新测序技术生成的大数据和先进的计算策略,如基因组装配与基因发现软件、统计建模与仿真、基因注释工具等,可以对微生物群进行准确分析(图1)。计算机和测序技术的蓬勃发展,极大地促进了整个人类微生物学领域的发展。
综合MetaHit和人类微生物组计划的数据可以获得迄今为止人体相关微生物最全面的信息。这些研究汇总数据表明,从人体中共分离出2172种微生物,分为12个不同的门,其中93.5%属于Proteobacteria、Firmicutes、Actinomycetes和Bacteroidetes。在人体中,有386种已鉴定的菌种为严格厌氧,通常定殖在口腔和胃肠道等黏膜区域[6]。目前已知Actinomycetes、Firmicutes、Facteroidetes和Proteobacteria在人体胃中占主导地位,而Proteobacteria、Firmicutes常见于肠道。共生肠道菌群具有多样性、稳定性、抗性和恢复力强等特点,而非共生肠道菌群相对丰度较低,并缺乏共生性和多样性[7]。
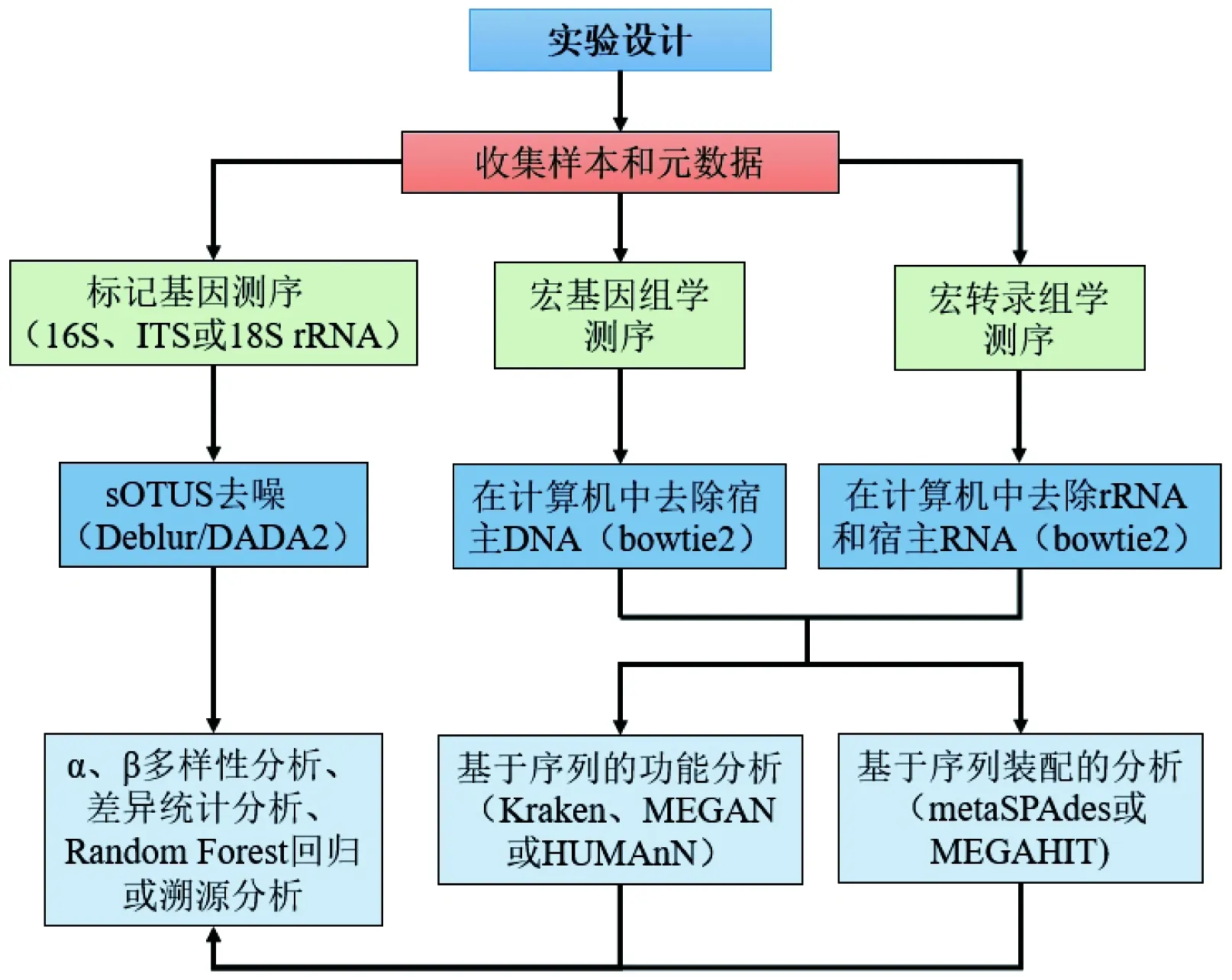
图1 16S rRNA、宏基因组学和宏转录组学测序分析流程
胃肠道微生物群的多样性并不像身体其他部位,如皮肤和口腔那样丰富,并且胃肠道菌群显示出高度的功能冗余[8]。有研究通过249个新测序和1018个已发表样本组合鉴定出近千万个基因,从而获得了人类肠道微生物组的功能目录。这项研究分析了不同国家人群肠道的菌群特征,证明肠道微生物群的组成由环境因素和宿主基因共同决定[9]。但不同组成的微生物群可能具有某种程度的功能冗余,以及相似的蛋白质或代谢产物谱。这一信息对于制定有关改善和塑造人类疾病中胃肠道微生物群的治疗策略至关重要。
2 胃部微生物与胃部疾病
人体胃部微生物群鉴定和分类的研究最初是基于经典的微生物体外培养技术和胃标本,包括黏膜和胃液。在健康状况下,人体胃部可以分离培养的菌属主要有Clostridium、Lactobacillus和Veillonella。随着高通量DNA焦磷酸测序、宏基因组测序和16S rRNA基因测序等先进的分子技术的出现,人们对于胃微生物组的生物多样性有了更充分的了解[10]。基于这些测序分析发现人体胃部在健康条件下的菌属主要有Prevotella、Streptococcus、Veillonella、Rothia、Pasteurellaceae、Fusobacterium、Actinomyces、Neisseria、Haemophilus和Porphyromonas,而且胃腔及胃体各部分的微生物群没有实质性的差异[11]。
虽然胃部微生物群结构组成的形成机制尚不明确,但普遍认为饮食、抗生素、益生菌、长期使用质子泵抑制剂(PPIs)或H2拮抗剂和H.pylori感染等多种因素都会改变胃部微环境[12](图2)。其中,长期使用PPI或H2拮抗剂会抑制胃酸分泌,对胃微生物群具有严重影响。早期研究报道,个体抗酸处理后胃内细菌会过度生长[13]。人体在补充维生素D3后胃部菌群多样性增加,但肠道微生物群不会受到影响,说明某些因素可能会优先影响胃部微生物群的组成,而不会干扰人体其他部位的微生态[14]。研究表明遗传背景似乎不会影响胃部微生物群的组成,同卵双胞胎之间的胃部微生物群组成与不相关的个体相比没有显著区别[15]。另一个可能调节胃部微生态的因素是宿主的免疫状态,因为微生物群的组成对人体免疫细胞有着重要影响,反之,宿主良好的免疫反应有助于调控胃部微生物群失调[16]。免疫抑制状态、抗生素治疗和胃液pH值大于4均可能引起胃部微生物群多样性的降低,其中免疫抑制与Lactobacillus过度生长和Prevotella、Fusobacterium丰度降低有关[17]。
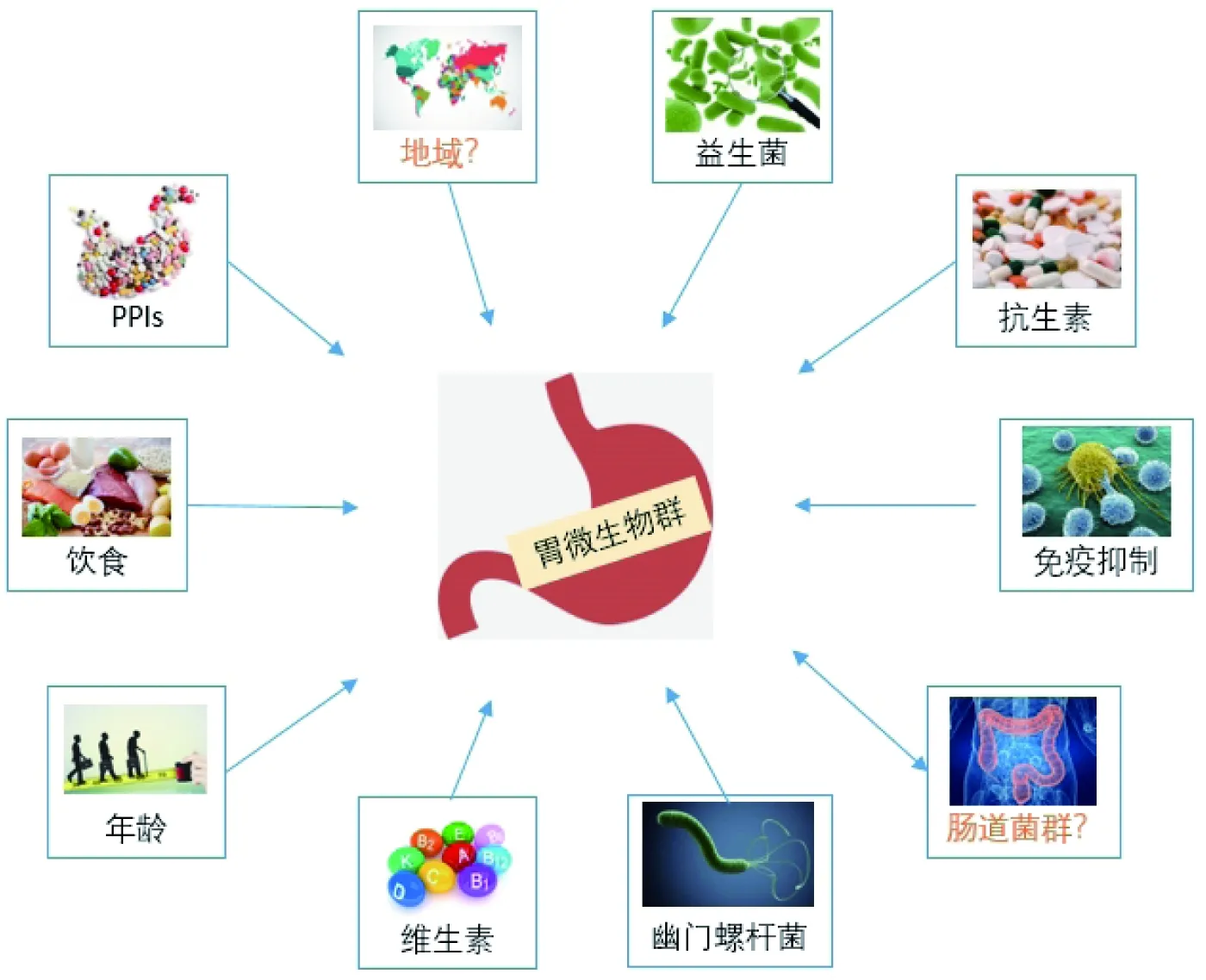
图2 胃微生物群的影响因素
幽门螺杆菌(Helicobacterpylori)是一种常见于人类胃部的细菌,是消化性溃疡、胃癌和黏膜相关淋巴组织(MALT)淋巴瘤的主要致病菌。胃癌是全球第四大常见癌症,其大多数病例发生在发展中国家。H.pylori感染是胃癌发生发展的最大危险因素,占全球胃癌的75%[18]。H.pylori具有多种毒力因子,其中细胞毒素相关基因A(CagA)、空泡细胞毒素(VacA)和外膜蛋白(OMPs)直接或间接与H.pylori的致癌性有关[19]。除了这些毒力因子外,胃部感染H.pylori所产生的慢性炎症也是促进胃癌发生发展的关键因素[20]。
H.pylori和胃微生物群之间的相互作用还没有完全阐明,但H.pylori的定殖可能诱发特定胃微生物群组成的变化,引起胃微生物群的失调。H.pylori感染阳性患者的胃微生物组与未感染个体的胃部微生物组有所不同,其胃部non-Helicobacter、Proteobacteria和Spirochetes丰度较高[21]。在有消化不良症状的儿童中,H.pylorie感染阴性患者的菌群多样性更高,主要类群有γ-Proteobacteria、β-Proteobacteria、Bacteroidia和Clostridia[22]。但目前还不清楚是H.pylori感染本身促进有害微生物的生长,还是微生物群失调引起黏膜或胃腔的变化,从而为H.pylori的定殖创造了有利条件,很有可能存在一种双向的相互作用。
H.pylori在胃癌发生中的作用毋庸置疑,然而有证据表明胃部微生物群中的其他微生物也可能与胃上皮细胞的癌变有关。胃癌组织具有独特的微生物谱,与正常胃组织相比胃癌组织的微生物群多样性有所降低,而根除H.pylori后菌群多样性可以恢复[23]。胃部Lactobacilluscoleohominis和Lachnospiraceae的增加,Porphyromonas、Neisseria和Streptococcussinensis的减少均可能与胃癌的发生具有相关性[24]。此外,一项对胃癌患者胃大部切除术前后胃微生物群的调查研究显示,胃癌患者胃部微生物群的多样性在手术后具有特异性的改变,表现为Proteobacteria和Actinobacteria数量减少以及Firmicutes和Bacteroidetes数量增加[25]。最近有研究发现在胃癌发生发展的不同阶段(从浅表性胃炎、萎缩性胃炎、肠上皮化生到胃癌),胃部微生物群出现显著失调,并且口腔微生物Peptostreptococcusstomatis、Streptococcusanginosus、Parvimonasmicra、Slackiaexigua和Dialisterpneumosintes相互影响逐渐形成致癌作用越来越强的特异性微生态,体现了口腔致病性菌群在胃癌的无创诊断中的潜在应用价值[26]。
血清胃蛋白酶原I与胃蛋白酶原II比值降低与上消化道微生物群多样性的降低有关,这一比值也与胃癌的发生具有相关性[27]。此外,胃癌患者的胃液中亚硝酸盐的含量高于健康对照组,可能是其胃内较多的厌氧菌促进了胃内亚硝酸盐的积累[28]。新的研究发现,与慢性胃炎相比,胃癌微生物群的物种多样性降低、H.pylori数量减少以及具有潜在遗传毒性的微生物比例升高,同时由PICRUSt功能预测得知胃癌微生物群硝酸盐还原酶和亚硝酸盐还原酶的功能增强[29]。
虽然我们对胃微生物群的认识有了很大的拓展,但目前对胃微生物群的研究大多局限于小群体中的个体,因此需要设计包括大量个体的纵向前瞻性研究,明确阐明胃微生物群在疾病发展中的作用。此外,还需要进一步研究抗炎药物、类固醇和免疫抑制剂在H.pylori存在或不存在的情况下对胃微生物群的影响。同时需要开发新方法,能够快速、准确识别并描述胃微生物群的形成与功能,例如,能够测量微生物代谢活性的方法将有助于开发适用于临床环境的微生物群检测技术。而了解胃癌发生发展过程中微生物群的动态,将为潜在的预防、诊断和治疗胃癌的临床应用开辟新的方向。
3 肠道微生物与肠道疾病
近十年来,微生物学研究的热潮为人们描绘出了肠道微生物的组成和某些功能的蓝图。人们在研究炎症性肠病(IBD)和结直肠癌(CRC)的肠道微生物群组成方面做出了巨大努力和重大进展,但是由于人类肠道微生物群的复杂性,许多微生物功能方面的问题仍未解决。
炎性肠病(IBD),包括克罗恩病和溃疡性结肠炎,在全世界范围内发病率逐渐增加[30]。IBD的特征是免疫介导的肠道慢性炎症,受遗传易感性和环境因素如饮食、抗生素的使用等影响[31]。临床发现抗生素可以治疗IBD,这与肠道菌群可能引起炎症反应的观点相一致[32]。有研究发现,克罗恩病患者的肠道微生态失调反映了炎症的存在及其严重程度,同时微生态失调与饮食和抗生素使用等因素有关联[33-34]。因此,肠道微生物群改变可能在IBD的早期出现并导致疾病的发生,而炎症在内的环境因素可能通过改变肠道的代谢条件进一步导致微生态失调。IBD患者微生物群会发生整体变化,但是某些特定病原体,如Enterobacteriaceae以及Mycobacteriumavium的亚种paratuberculosis可能是IBD的潜在病原体[35]。并且在溃疡性结肠炎患者肠道中可以分离出高侵袭致病性菌株Fusobacteriumnucleatum[36]。肠道微生物群可能诱导IBD发生,但目前为止的研究都只阐述了相关性而未证明因果关系。IBD中的微生态失调在很大程度上反映了复杂微生物群对肠道炎症等环境压力的响应,同时与炎症和病原体定殖相关的微生物代谢改变可能促进微生态失调。
肠道微生物群组成的变化导致的代谢产物改变,可能在IBD发病过程中起作用。代谢组学研究表明,健康个体和IBD患者的肠道微生物群之间有12%的代谢途径明显不同。具体而言,IBD患者微生物群中氨基酸合成和碳水化合物代谢途径减少,营养摄取和毒力的分泌途径增加[37]。生物信息学分析显示,与健康个体相比,IBD患者肠道微生物群中胆盐水解酶(BSH)的分泌显著降低, Firmicutes占比显著下降[38]。与IBD相关的另一种细菌代谢途径是某些碳水化合物经微生物发酵产生的短链脂肪酸(SCFAs)。Clostridia产生的SCFAs可以激活G蛋白偶联受体以及通过表观遗传效应增强肠黏膜中调节性T(Treg)细胞功能,从而促进免疫耐受的恢复并减少结肠炎的发生[39]。
粪菌移植(FMT)成功治疗艰难梭菌感染激起了人们使用FMT治疗IBD的研究兴趣[40]。然而,目前为止FMT治疗IBD的临床结果不一致,可能是因为与艰难梭菌感染引起的结肠炎相比IBD的复杂性更高[41-43]。在使用安慰剂作为对照的两项治疗溃疡性结肠炎的随机试验表明,在缓解溃疡性结肠炎成年患者的临床症状方面,FMT明显比安慰剂更有效[44-45]。但是,在治疗肠应激综合征(IBS)的随机双盲临床试验中,FMT没有表现出很好的疗效,同时FMT组出现了不良反应[46]。因此,FMT(短期和长期)的安全性和持久性、对免疫抑制患者的影响、最有效的管理方式以及如何选择合适的供体和受试者等方面仍然存在疑问。为了更好地确定FMT在IBD治疗中的作用,必须进行更大规模临床随机对照试验。
结肠直肠癌(CRC)常与肿瘤及其邻近黏膜处的微生物群失调有关,但CRC进展中的优势菌种仍不清楚。研究分析发现CRC患者粪便中菌群多样性降低,其中纤维发酵相关菌类Clostridia减少,口腔共生Fusobacteriumnucleatum和Porphyromonas增加[47]。有研究表明F.nucleatum通过干扰TLR4和MyD88信号通路使CRC患者产生对抗肿瘤药物Oxaliplatin和5-FU的耐药性并更容易复发,F.nucleatum还可以靶向microRNA激活自噬途径从而改变化疗反应[48]。F.nucleatum感染CRC细胞可增加其增殖率和侵袭活性,同时F.nucleatum还具有诱导小鼠产生异种移植瘤的潜能[49]。某些Escherichiacoli可以促进肠道炎症的发生,产生具有致癌性的毒素如大肠杆菌素[50]。而黏膜相关E.coli在CRC组织中更为普遍,可能与肿瘤分期和预后有关[51]。宏基因组研究发现E.coli和F.nucleatum在CRC发生发展中的作用不尽相同[52]。例如,临床CRC患者的F.nucleatum相关分离物未能诱发小鼠炎症和癌症,而产大肠杆菌素E.coli却能促进ApcMin/+;Il10-/-小鼠肿瘤发生,但该临床表现可能受F.nucleatum定殖程度和肠道肿瘤位置影响[53]。Bacteroidesfragilis构成人体共生微生物群的1%~2%,其衍生毒素(BFT)可以引起炎症性腹泻和炎症相关肿瘤的发生[54]。分泌BFT的肠毒性B.fragilis(ETBF)可由炎症性Th17细胞驱动,诱导ApcMin/+小鼠产生结肠炎和结肠肿瘤[55]。使用头孢西丁治疗可完全清除ETBF的定殖,从而减少小鼠IL-17A表达和结肠肿瘤发生[56]。BFT还可能上调精胺氧化酶产生活性氧并引起DNA损伤,进而导致炎症和肿瘤发生[57]。多项动物实验证明F.nucleatum、产大肠杆菌素E.coli和肠毒性B.fragilis及其代谢产物与CRC的发生发展具有相关性,但其因果关系还需进一步的临床研究。
研究发现Parvimonasmicra和Solobacteriummoreii等也与CRC发生相关,使用肠道微生物群中20个菌种的基因和两种酶基因(F.nucleatum的丁酰辅酶A脱氢酶和P.micra的RNA聚合酶亚基β)可以显著地区分出CRC患者[58]。而使用F.nucleatum、Bacteroidesclarus、Roseburiaintestinalis和Clostridiumhathewayi构建模型预测受试人群中CRC患者,其敏感性>90%,特异性>80%[59]。结肠肿瘤与癌旁黏膜间的菌群失调比例相似,某些特征菌群组成甚至存在重叠,说明结肠在癌症环境中已经出现相当程度的微生物群失调,因此CRC的粪便评估能充分反映黏膜活检的微生物组成[60]。上述研究表明,通过检测人体粪便中某些菌株及其衍生因子可作为CRC预防及诊断的筛选工具。
肠道微生物多样性降低是许多肠道和肠外疾病的特征,共生微生物群中某一类细菌的增加或减少以及特定菌种丰度改变都可能促进炎症以及肿瘤的发生,但其相关机制仍需进一步研究。值得注意的是,IBD患者的肠道微生物群表现出与CRC患者相似的组成改变,看来不仅是炎症,菌群失调也可能是CRC和IBD发生的关联因素之一[61]。因此,深入了解宿主微生物群的互利共生性和环境信号引起的干扰,对开发肠道疾病的新型治疗方法具有重要意义。
4 总结
对人体胃肠道微生物群的认识在快速发展,目前宏基因组关联分析可以确定微生物群组成的主要影响因素,并将单个微生物在物种水平上与宿主疾病、生活方式和生理学联系起来。而宏转录组学方法的应用也极大地促进了我们对胃肠道微生物群组成及其代谢产物影响人类健康和疾病相关机制的理解。胃肠道微生物群与宿主之间存在着密切的共生关系,消化系统的众多疾病环境中,均可以观察到其微生物群组成的紊乱。无论胃肠道微生物群失调是疾病的起因还是结果,都可能加剧疾病的进展,并影响相关的治疗策略。
随着对胃肠道微生物群组成功能认识的不断深入,通过调控微生物群组成来改善患者疾病状况的治疗方法受到广泛关注。然而目前对于健康微生物组仍未达成共识,有效、精准的微生物治疗手段还处于起步阶段,还需要推动更多更大规模的纵向和介入性研究,使用更新的方法学,包括多种基因组技术和新的体外方法,同时还要更多地关注微生物组本身的相互作用。这些研究将进一步加深对胃肠道微生物组的理解,阐明其致病机制,开发更安全有效、有针对性的肠道菌群疗法,从而更好地预防和治疗疾病。
