Pharmacogenetics of the systemic treatment in advanced hepatocellular carcinoma
2019-08-20ElenaDeMattiaErikaCecchinMichelaGuardascioneLuisaFoltranTaniaDiRaimoFrancescoAngeliniMarioAndreaGiuseppeToffoli
Elena De Mattia, Erika Cecchin, Michela Guardascione, Luisa Foltran, Tania Di Raimo, Francesco Angelini,Mario D’Andrea, Giuseppe Toffoli
Abstract Hepatocellular carcinoma (HCC) accounts for the majority of primary liver cancers. To date, most patients with HCC are diagnosed at an advanced tumor stage, excluding them from potentially curative therapies (i.e., resection, liver transplantation, percutaneous ablation). Treatments with palliative intent include chemoembolization and systemic therapy. Among systemic treatments, the small-molecule multikinase inhibitor sorafenib has been the only systemic treatment available for advanced HCC over 10 years. More recently, other smallmolecule multikinase inhibitors (e.g., regorafenib, lenvatinib, cabozantinib) have been approved for HCC treatment. The promising immune checkpoint inhibitors(e.g., nivolumab, pembrolizumab) are still under investigation in Europe while in the US nivolumab has already been approved by FDA in sorafenib refractory or resistant patients. Other molecules, such as the selective CDΚ4/6inhibitors (e.g.,palbociclib, ribociclib), are in earlier stages of clinical development, and the c-MET inhibitor tivantinib did not show positive results in a phase III study.However, even if the introduction of targeted agents has led to great advances in patient response and survival with an acceptable toxicity profile, a remarkable inter-individual heterogeneity in therapy outcome persists and constitutes a significant problem in disease management. Thus, the identification of biomarkers that predict which patients will benefit from a specific intervention could significantly affect decision-making and therapy planning. Germ-line distribute, remix, adapt, build upon this work non-commercially,and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See:http://creativecommons.org/licen ses/by-nc/4.0/
Key words: Hepatocellular carcinoma; Pharmacogenetics; Genetic markers; Sorafenib;Regorafenib; Immune checkpoint inhibitors; Cytochromes; UDP glucuronosyltransferase 1A
INTRODUCTION
Liver cancer incidence is approximately 850000 new cases per year, and about 90% of liver tumors are hepatocellular carcinoma (HCC)[1]. The dominant risk factors for HCC vary worldwide. For most countries in Asia and Africa, hepatitis B virus infection and aflatoxin B1 exposure are the major risk factors. In contrast, hepatitis C virus infection, alcoholism, and metabolic syndrome play more important roles in other areas in the world[2]. The Barcelona Clinic Liver Cancer (BCLC) staging is the main clinical classification that stratifies patients from A to C stages, according to prognosis, to inform treatment decisions. Early-stage cancers are potentially suitable for therapies with curative intent such as surgical resection, liver transplantation, or local ablation. Chemoembolization and systemic therapy represent the only therapeutic options for intermediate or advanced HCC[1].
Surgical resection is the standard option for patients with solitary HCC at BCLC A stage. Other criteria for selecting the best surgical candidates are absence of portal hypertension along with well-preserved liver function. A surgical strategy is associated with 5-year survival rates of 70%, and adjuvant therapies have yet to show a survival advantage[1]. Liver transplantation is the best option for BCLC A tumors with respect to Milan criteria (single tumor ≤ 5 cm or up to three nodules ≤ 3 cm in size and no vascular invasion)[3]. Furthermore, local ablation with radiofrequency represents a good alternative to surgery in patients with single tumors < 2 cm[4];however, no randomized clinical trials (RCTs) have been conducted to specifically address whether ablation is non inferior to surgery[5].
For patients with HCC at BCLC B stage, transarterial chemoembolization (TACE) is recommended based on results from RCTs and a systematic review showing survival benefits with TACE as compared with the best supportive care[6]; more recently TACE was found to give an objective response of 52.5%[7]. Better selection of candidates and improvement in the procedure, such as supra-selective embolization and the use of drug-eluting beads, have led to median survival times beyond 40 mo in referral centers[1]. Radioembolization is an alternative embolization approach with a favorable safety and efficacy profile, but well-designed, properly powered RCTs are still needed to demonstrate a real benefit[1].
In advanced HCC or in intermediate HCC when chemoembolization is no longer indicated, systemic treatment is the standard therapy. Conventional chemotherapeutic agents (e.g., doxorubicin, fluoropyrimidines, platinum derivates,irinotecan) are minimally effective in HCC, with significant toxicity, and do not improve patient survival[8-10]. HCC is also rarely amenable to radiation therapy[10].
Targeted agents based on an improved molecular characterization of HCC have opened a new era for the treatment of patients with HCC (Figure 1). A number of small-molecule tyrosine kinase inhibitors and immune checkpoint inhibitors have demonstrated some survival benefit in intermediate/advanced disease (BCLC B-C);more recently, preliminary promising data are emerging on the use of CDΚ4/6inhibitors[11]. At present, the approved drugs in Europe for advanced HCC indication are the small-molecule multikinase kinase inhibitors sorafenib, lenvatinib,regorafenib, and cabozantinib. In particular, sorafenib and lenvatinib are approved as first-line therapy and regorafenib and cabozantinib in patients who have progressed or are intolerant to sorafenib. Other molecules, such as the immune checkpoint inhibitors (e.g., nivolumab, pembrolizumab) and selective CDΚ4/6inhibitors (e.g.,palbociclib, ribociclib), are still under investigation in the HCC setting in Europe while in the US nivolumab received accelerated approval for HCC patients previously treated with sorafenib. Among other molecules tested, the c-MET inhibitor tivantinib has not shown positive results[12].
Based on the results of major studies conducted to date, several unmet needs persist in the management of intermediate/advanced HCC that might be addressed through new therapies and biomarkers for therapy stratification and a patient-tailored approach. In this context, genetic polymorphisms, with their well-established role in liver carcinogenesis[13,14], could be important and contribute, in combination with clinical and molecular parameters, to predicting HCC therapy outcomes for efficacy and for toxicity risk. The aim of this review is to critically report and discuss current literature on the effect of germ-line variants as predictive markers of HCC systemic therapy outcome and how they can aid in stratifying patients according to toxicity risk, as well as the likelihood of benefit from administration of specific anti-tumor agents.
SYSTEMIC TREATMENT OF ADVANCED HCC
The phase III SHARP trial evaluating sorafenib in previously untreated patients with advanced HCC reported a median overall survival (OS) of 10.7 mo for the sorafenibtreated group compared to 7.9 mo in patients who received placebo[15]. The most common adverse effects observed in the trial included fatigue, hand-foot skin reaction (HFSR), alopecia, gastrointestinal, and liver dysfunction. A number of studies have investigated the role of clinical and/or biological markers in HCC patients treated with sorafenib[15,16]. Results from the SHARP trial showed that baseline alpha fetoprotein plasma levels > 200 ng/mL had a negative impact on OS, a finding that has been recently confirmed in a pooled analysis[17]. A recent meta-analysis demonstrated that the occurrence of sorafenib-related side effects (e.g., hypertension,skin toxicities, and diarrhea) is associated with a better OS in sorafenib-treated HCC patients[18]. In addition to the abovementioned markers, other clinical parameters have been evaluated, such as macroscopic vascular invasion, BCLC stage and etiology of cirrhosis[17], and Child-Pugh subgroups[19]. Some biological markers have been also suggested as potentially related to sorafenib outcome. For instance, in the SHARP trial[15], baseline angiopoietin-2 (Ang-2) and vascular endothelial growth factor(VEGF)-A plasma levels independently predicted survival in the entire patient population and in the placebo cohort; conversely, none of the tested biomarkers significantly predicted response to sorafenib[15]. Additionally, high insulin-like growth factor 1 pre-treatment levels are associated with better progression-free survival (PFS)and OS in patients with advanced HCC receiving first-line antiangiogenic therapy[20].More recently, a study recruiting 80 HCC patients prospectively treated with sorafenib showed that independent risk factors for poor OS were high serum concentration of Ang-2 and hepatocyte growth factor (HGF) as well as poor performance status before treatment[21].
In patients who tolerated but progressed on sorafenib, the other multikinase inhibitor regorafenib has been reported to provide an OS benefit compared with placebo of 10.6 mo vs 7.8 mo. The most common grade 3 or 4 treatment-related events were hypertension, HFSR, fatigue, and diarrhea[22]. Preliminary data on potential biomarkers of response to regorafenib in patients with HCC have been recently published. Particularly, a study involving a large cohort of patients enrolled in the phase III RESORCE trial showed a significant association of OS with plasma concentrations of some proteins involved in inflammation and/or HCC pathogenesis as well as a number of plasma miRNAs. In addition, a somatic profile of tumor tissues was described that suggested a potential mutational pattern associated with response to regorafenib[23].
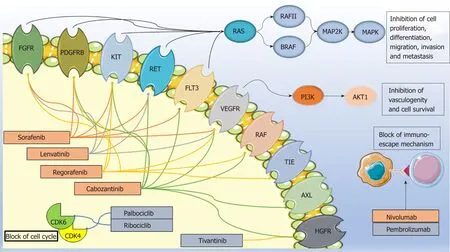
Figure 1 Mechanisms of action of the drugs covered in the text. Approved drugs are in the orange box while those under approval are in the grey box. Image created with Servier Medical Art (https://smart.servier.com/). FGFR1: Fibroblast growth factor receptor; PDGFR: Platelet-derived growth factor receptor; FLT3: Fmsrelated tyrosine kinase 3; VEGFR: Vascular endothelial growth factor receptor; TIE: Tyrosine kinase with immunoglobulin-like and EGF-like domains; HGFR:Hepatocyte growth factor receptor; PD1: Programmed cell death protein-1; PDL1/2: programmed cell death protein ligand 1/2; CDK: Cyclin-dependent kinases.
More recently, a phase III trial comparing lenvatinib to sorafenib in the first-line setting showed non-inferiority of lenvatinib to sorafenib for the primary endpoint OS and statistically significant improvement for secondary end-point PFS. The most common any-grade adverse events described for lenvatinib were hypertension,diarrhea, and appetite and weight reduction. In addition, there were fewer dermatological adverse events but more hypertension for lenvatinib compared to sorafenib[24]. Finally, the small-molecule multikinase inhibitor cabozantinib was associated with longer OS than placebo in a phase III trial involving patients already treated for advanced disease. In that study, incidence of grade 3 or 4 adverse events was higher (predominantly grade 3) in the cabozantinib arm, including palmar-plantar erythrodysesthesia and HFSR, hypertension, increased aspartate aminotransferase (AST), fatigue, and diarrhea[25].
Other molecules not yet approved in Europe for the treatment of liver cancer are under investigation in the HCC setting, with promising preliminary results.Particularly, the novel class of immune checkpoint inhibitors has demonstrated significantly improved survival outcomes for patients with HCC. A phase I/II study trial investigated the role of the immunotherapeutic agent nivolumab in patients whose disease progressed while receiving at least one previous line of systemic therapy, including sorafenib, or who were intolerant to sorafenib. In this trial 262 eligible patients were treated, 48 in the dose-escalation phase and 214 in the doseexpansion phase. During dose escalation, 12 (25%) patients had grade 3 or 4 adverse events while 3 (6%) patients had serious adverse events (i.e., pemphigoid, adrenal insufficiency, liver disorder); the objective response rate was 15% (95%CI: 6%-28%).For dose expansion, the objective response rate was 20% (95%CI: 15%-26%) with nivolumab 3 mg/kg[26]. Based on the results of this study, the U.S. Food and Drug Administration (FDA) granted accelerated approval of nivolumab on September 2017.A phase III randomized trial of first-line nivolumab compared with sorafenib is ongoing (ClinicalTrials.gov Identifier: NCT02576509). Another phase II trial focused on another immune checkpoint inhibitor, pembrolizumab, in patients with HCC pretreated with sorafenib. These results showed that pembrolizumab was effective and tolerable, with fatigue and increased AST as the most frequent adverse events[27].Several phase II/III clinical trials with immunotherapeutic agents are currently recruiting HCC patients worldwide. One is an ongoing phase III randomized, activecontrolled trial to evaluate the safety and efficacy of lenvatinib in combination with pembrolizumab compared with lenvatinib plus placebo in first-line therapy for advanced HCC (ClinicalTrials.gov Identifier: NCT03713593). A phase II trial with sorafenib and nivolumab as first-line therapy is also in progress (ClinicalTrials.gov Identifier: NCT03439891).
Among the molecules at an earlier stage of clinical development in HCC, the selective CDΚ4/6 inhibitors stand out. In an early trial, palbociclib demonstrated activity in patients with advanced HCC after failure of first-line sorafenib. This trial enrolled 21 patients, 4 being non-evaluable. In evaluable patients median OS was 19 wk and median time to progression was 24 wk; prolonged stability was seen in 3 patients. The most common grade 3 or 4 adverse events were neutropenia and thrombocytopenia, and non-serious adverse events were anemia, pain, ascites, and fatigue[11]. A phase Ib/II study of another CDΚ4/6 inhibitor, ribociclib, in association with chemoembolization in advanced HCC is currently recruiting patients(ClinicalTrials.gov Identifier: NCT02524119).
Another molecule under investigation that should be cited for completeness is tivantinib, a selective inhibitor of the proto-oncogene MET, belonging to the class of the small-molecule kinase inhibitors. A phase II randomized trial evaluated the administration of tivantinib as the second-line therapy for patients with HCC. The study showed improved PFS for tivantinib compared with placebo in a subset of patients with high MET expression tumors, and the most common grade 3 or worse adverse events in the tivantinib group were neutropenia and anemia[28]. On 13 November 2013, orphan designation (EU/3/13/1202) was granted for tivantinib for the treatment of HCC in patients whose disease has stopped responding or is resistant to sorafenib. However, a subsequent phase III trial evaluating the use of tivantinib for second-line treatment of MET-high expressing advanced HCC showed no OS improvement for tivantinib compared with placebo in patients previously treated with sorafenib[12].
PHARMACOGENETICS OF APPROVED DRUGS
Sorafenib
Sorafenib (NEXAVAR®) is an orally administered multi-targeted tyrosine kinase inhibitor. This small molecule inhibits a number of serine/threonine and tyrosine kinases [e.g., VEGF receptors (VEGFR1-3), platelet-derived growth factor receptor(PDGFR), fibroblast growth factor receptor 1 (FGFR1), ΚIT proto-oncogene receptor tyrosine kinase (ΚIT), ret proto-oncogene (RET], and fms-related tyrosine kinase 3(FLT3)] and downstream oncogenic Raf signaling players (e.g., Raf-1 and B-Raf). Thus,it affects multiple tumor-related signaling pathways, such as those involved in angiogenesis, tumor proliferation, and cell apoptosis[29,30]. Although survival improvement has been achieved with this targeted agent, only a limited number of patients have experienced a real and long-term benefit. Moreover, a high resistance rate and some significant and expensive toxicities further restrict the advantages of sorafenib therapy and constitute a crucial problem in HCC management.
In recent years, some pharmacogenetic studies have focused on identifying genetic markers that could predict risk for severe adverse events (Table 1) and discriminate sorafenib-responsive patients from non-responders (Table 2). Details regarding the pharmacogenetic panel analyzed, the study population (e.g., sample size, ethnicity)and therapy (e.g., dose and schedule) characteristics, the clinical end-points evaluated along with the main findings (e.g., statistical results) of the studies are shown in Tables 1 and 2.

Table 1 Published works on germ-line variants and pharmacokinetic and toxicity profiles of sorafenib in hepatocellular carcinoma patients Ref.][32][34][35][36][42 Main findings Rat models: CYP3A4*1 (rs2740574) and CYP3A5*3 (rs776746) were associated with the lowest (8 ± 2.5 ng/mL) and highest (67 ± 4.8 ng/mL) levels of sorafenib plasma concentration, respectively (P < 0.001). CYP3A5*3 correlated with the most severe liver [change in ALT and AST blood concentration [(IU/L) over time] and renal injury (change in BUN [nmol/L) and Cr [umol/L] blood concentration [IU/L]over time) and CYP3A4*1 with the least severe injury (P < 0.001)Clinical setting: CYP3A5*3 was associated with increased severe hepatic toxicity (change in ALT and AST blood concentration [IU/L]over time, P < 0.001)UGT1A9 rs17868320-T allele was associated with increased grade ≥ 2 diarrhea (OR: 14.33, P = 0.015, multivariate analysis)ABCG2 rs2622604-TT genotype exhibited a greater exposure compared to the CC (sorafenib AUC, 131.8 vs 82.4 mg/L.h, P = 0.093 univariate analysis, not confirmed in multivariate)UGT1A1*28 (rs8175347) was associated with increased risk of acute hyperbilirubinemia (*28/*28 vs other, OR:5.413, P = 0.016) and of interrupting treatment (*28 vs other, OR: 3.397, P = 0.002) by multivariate analysis. The *28 allele also showed a trend towards a higher risk for any toxicity at grade 3 or higher (P = 0.088)SLCO1B1*1b (rs2306283-G) allele was associated with inferior risk of diarrhea (OR: 0.125, P = 0.007) and increased risk of hyperbilirubinemia (OR: 1.230, P = 0.002), and the SLCO1B1*5 (rs4149056-C) allele with higher risk of thrombocytopenia (OR: 4.219, P =0.045) in univariate but not multivariate analysis No SNP was associated with OS or PFS VEGFA 1991-CC (OR: 45.68, P = 0.011), TNFA rs1800629-GG (OR: 44.06, P = 0.023), and UGT1A9 rs7574296-AA (OR: 18.717, P = 0.015)were independent risk factors for the development of high-grade HFSR (multivariate analysis).ABCB1 rs2032582 (CT: 0.7 ± 0.6 kg/L vs TT:2.3 ± 2.2 kg/L, P = 0.035), ABCG2 rs2231137 (AG: 0.8 ± 0.4 kg/L vs GG: 1.4 ± 1.5 kg/L, P = 0.02),ABCG2 rs2231142 (CA: 0.5 ± 0.5 kg/L vs CC: 1.4 ± 1.4 kg/L, P = 0.007) heterozygous genotypes were associated with the lowest sorafenib plasma levels Clinical endpoint Toxicity PΚ Toxicity Toxicity PFS OS Toxicity PΚ Therapy Sorafenib Sorafenib 400 or 200 mg,twice daily Sorafenib Sorafenib 400 mg twice daily in combination with TACE Sorafenib 400 mg twice daily Study population Advanced HCC(n = 51)(Chinese)Aflatoxin-induced HCC rat models(n = 105)Advanced solid cancer(n = 54; 37% HCC)(whites)Advanced solid cancer(n = 114; 87% HCC)(mainly whites)Intermediate stage HCC (n = 59)Advanced HCC(n = 47)(Caucasians)Pharmacogenetic panel CYP3A4*1, CYP3A5*3,CYP2C19*2,CYP2D6*10 9 SNPs in CYP3A5,UGT1A9, ABCB1,ABCG2 8 SNPs in SLCO1B1,SLCO1B3, ABCC2,ABCG2, UGT1A1,UGT1A9 49 SNPs in UGT1A9,UGT1A1, CYP3A4,CYP2B6, TNFA,VEGFA, IGF2, HIF1A (Κorean)5 SNPs in ABCB1,ABCG2 ALT: Alanine aminotransferase; AST: Aspartate aminotransferase; AUC: Area under the curve; BUN: Blood urea nitrogen; Cr: Creatinin; HCC: Hepatocellular carcinoma; HFSR: Hand-foot skin reaction; OR: Odds ratio; OS:overall survival; PFS: Progression-free survival; PΚ: Pharmacokinetic; SNP: Single nucleotide polymorphism; vs: Versus.

Table 2 Published works on germ-line variants and response to sorafenib in hepatocellular carcinoma patients Ref.][48][50(rs2071559-C, rs2305948-C) alleles were associated with longer PFS and OS Univariate analysis Main findings VEGFA (rs25648-C, rs833061-T, rs699947-C, rs2010963-C), VEGFC (rs4604006-T), VEGFR1 (rs664393-G), and VEGFR2 VEGFA rs2010963-C allele (PFS, 6.9 mo vs 4.0 mo, HR: 0.25, p = 0.0376; OS, 17.0 vs 9.3 mo, HR: 0.28, P = 0.0201), VEGFC rs4604006-T allele (PFS, 10.1 mo vs 4.3 mo, HR: 0.22, p = 0.004; OS, 22.0 mo vs 13.0 mo, HR: 0.25, P = 0.04) and BCLC C stage 10.7 mo, HR: 0.36, P = 0.0015) were independent prognostic factors Multivariate analysis(PFS, 7.6 mo vs 4.5 mo, HR: 0.17, P = 0.0163; OS, 21.0 mo predicting PFS and OS Patients with both the favorable alleles of VEGFA rs2010963 and VEGFC rs4604006 showed improved PFS and OS compared to those with only one or none (PFS: P = 0.0004; two favorable alleles: 11.4 mo, one favorable and one unfavorable: 5.6 mo,two unfavorable: 3.4 mo; OS: P = 0.0001, two favorable alleles: 22.7 mo, one favorable and one unfavorable, 15.1 mo, two unfavorable, 8.8 mo)VEGFA rs2010963-C (P = 0.0343) and VEGFC rs4604006-T (P = 0.0028) alleles were also associated with a better objective response Univariate analysis VEGFR2 rs1870377-AA (5.8 mo vs 4.0 mo, P = 0.001) and rs2305948-AA (5.8 mo vs 4.5 mo, P = 0.016) genotypes were associated with longer TTP VEGFR2 rs1870377-AA genotype (15.0 mo vs 9.6 mo, P = 0.001) and rs2071559-T allele (13.0 mo vs 9.0 mo, P = 0.007) were associated with longer OS VEGFR2 rs1870377-AA (P = 0.011) and rs2305948-AA (P = 0.047) genotypes were associated with a better response Multivariate analysis Major vascular invasion (HR: 2.51, P = 0.021) and VEGFR2 rs1870377-AA (HR: 0.68, P = 0.005) were independent factors in TTP; performance status (HR: 2.36, P = 0.017), VEGFR2 rs1870377-AA (HR: 0.35, P = 0.003) and rs2071559-CC (HR: 2.25, P =0.036) were independent factors in OS Clinical endpoint PFS OS Objective Response TTP OS Response Therapy Sorafenib 400 mg, twice daily First-line sorafenib 400, mg twice daily Study population Advanced or intermediatestage HCC (n =148)(whites)(ALICE-1)Advanced HCC(n = 78)(Chinese)Pharmacogenetic panel 17 SNPs in VEGFA, VEGFC, FLT1(VEGFR1), ΚDR (VEGFR2), FLT4 (VEGF3)18 SNPs in ΚDR (VEGFR2)
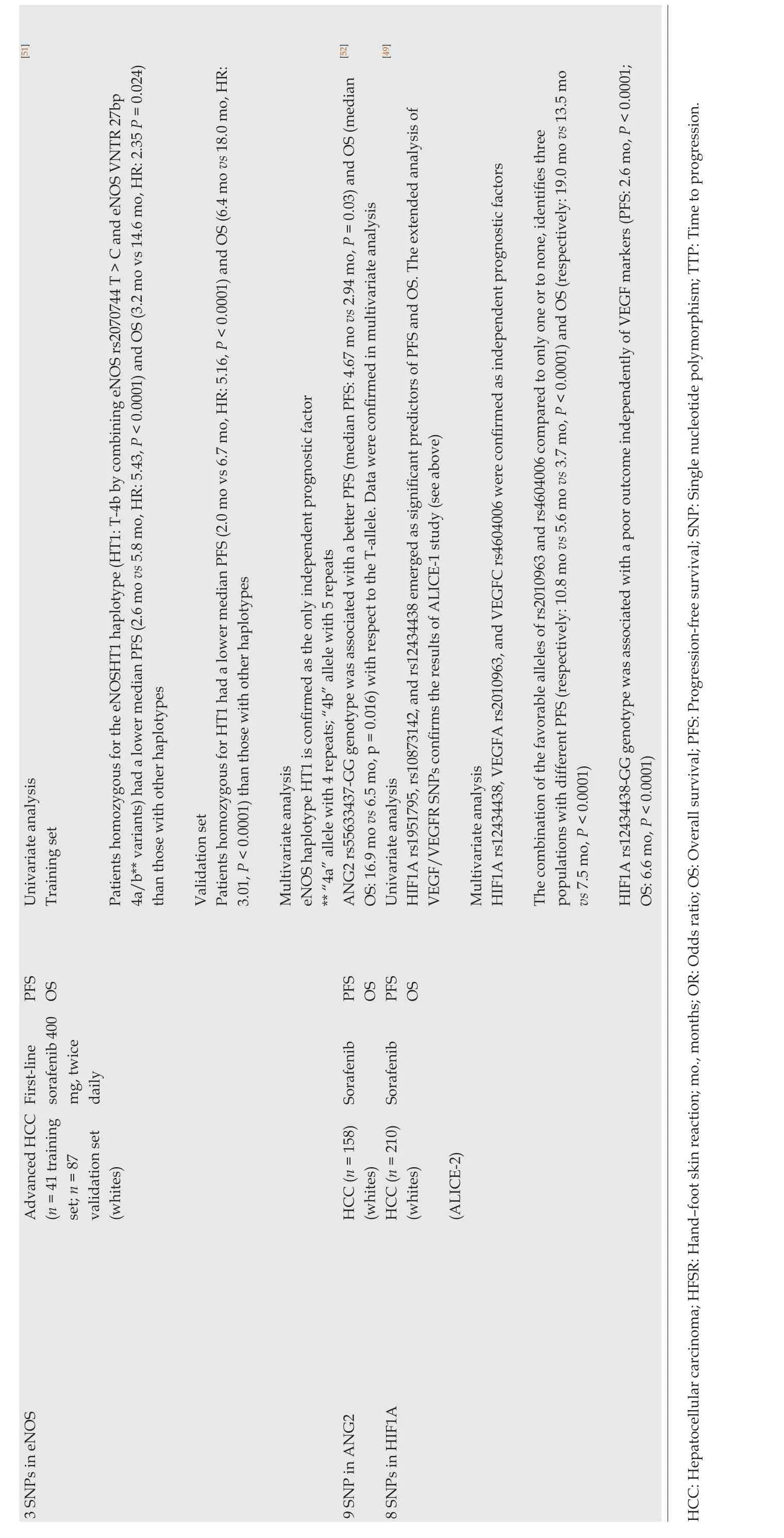
][51][52][49 Univariate analysis Training set Patients homozygous for the eNOSHT1 haplotype (HT1: T-4b by combining eNOS rs2070744 T > C and eNOS VNTR 27bp 4a/b** variants) had a lower median PFS (2.6 mo vs 5.8 mo, HR: 5.43, P < 0.0001) and OS (3.2 mo vs 14.6 mo, HR: 2.35 P = 0.024)than those with other haplotypes Validation set Patients homozygous for HT1 had a lower median PFS (2.0 mo vs 6.7 mo, HR: 5.16, P < 0.0001) and OS (6.4 mo vs 18.0 mo, HR:3.01, P < 0.0001) than those with other haplotypes Multivariate analysis eNOS haplotype HT1 is confirmed as the only independent prognostic factor** “4a” allele with 4 repeats; “4b” allele with 5 repeats ANG2 rs55633437-GG genotype was associated with a better PFS (median PFS: 4.67 mo vs 2.94 mo, P = 0.03) and OS (median OS: 16.9 mo vs 6.5 mo, p = 0.016) with respect to the T-allele. Data were confirmed in multivariate analysis Univariate analysis HIF1A rs1951795, rs10873142, and rs12434438 emerged as significant predictors of PFS and OS. The extended analysis of VEGF/VEGFR SNPs confirms the results of ALICE-1 study (see above)Multivariate analysis HIF1A rs12434438, VEGFA rs2010963, and VEGFC rs4604006 were confirmed as independent prognostic factors The combination of the favorable alleles of rs2010963 and rs4604006 compared to only one or to none, identifies three populations with different PFS (respectively: 10.8 mo vs 5.6 mo vs 3.7 mo, P < 0.0001) and OS (respectively: 19.0 mo vs 13.5 mo vs 7.5 mo, P < 0.0001)HIF1A rs12434438-GG genotype was associated with a poor outcome independently of VEGF markers (PFS: 2.6 mo, P < 0.0001;OS: 6.6 mo, P < 0.0001)PFS OS PFS OS PFS OS Advanced HCC First-line sorafenib 400 mg, twice daily(n = 41 training set; n = 87 validation set(whites)HCC (n = 158) Sorafenib(whites)HCC (n = 210) Sorafenib(whites)(ALICE-2)3 SNPs in eNOS 9 SNP in ANG2 8 SNPs in HIF1A HCC: Hepatocellular carcinoma; HFSR: Hand-foot skin reaction; mo., months; OR: Odds ratio; OS: Overall survival; PFS: Progression-free survival; SNP: Single nucleotide polymorphism; TTP: Time to progression.

Table 3 Published works on germ-line variants and pharmacokinetics, toxicity, and efficacy of regorafenibin solid cancer patients Ref.][60][61][57][63][62 Main findings SLCO1B1*1b (rs2306283-G) allele was associated with lower incidences of increased grade ≥ 2 AST (8% vs 42%, P = 0.03) and anemia (16% vs 50%, P= 0.048). A similar tendency was observed for the incidence of increased grade ≥ 2 ALT (4% vs 25%, P = 0.09) and total bilirubin (12% vs 25%, P = 0.37).ABCG2 rs2231142 was associated with different blood platelet counts (Plt, CC: 29.4 ± 10.7 *104/μL, CA + AA: 21.4 ± 11.3*104/μL, P = 0.03)SLCO1B1*5 (rs4149056-C) allele [17.3 (ng/mL)/mg vs 11.5 (ng/mL)/mg, p = 0.167] and SLCO1B1*1b (rs2306283-G/rs4149056-T) non-carriers [14.0(ng/mL)/mg vs 12.1 (ng/mL)/mg, P = 0.226] demonstrated a tendency toward higher concentration-to-dose ratio Drug concentrations were higher in the group with grade ≥ 2 total bilirubin elevation (3.45 µg/mL vs 1.76 µg/mL, P = 0.01) and thrombocytopenia(3.45 µg/mL vs 1.76 µg/mL, p = 0.02) compared with the group with grades 0-1 UGT1A9*22 (rs383204-T10) allele was associated with acute hepatitis (descriptive study)Univariate analysis VEGFA rs2010963-CC vs to CG + GG genotype was associated with both longer OS (9.0 mo vs 6.5 mo, HR: 0.52, P = 0.049) and PFS (2.2 mo vs 1.8 mo,HR: 0.49, P = 0.0038). The same genotype was also associated with better DCR (progression rate, 47% vs 74%, P = 0.02)VEGFR2 rs1870377 (TT: 1.97 mo, AT: 1.84 mo, AA: 1.12 mo; P = 0.0061) and VEGFR3 rs307805 (AA: 1.91 mo, AG: 2.07 mo, GG: 2.3 mo; P = 0.0492)were associated with OS only VEGFR1 rs664393 was associated with PFS only (CC: 2.01 mo, CT: 1.84 mo, TT: 1.48 mo; P < 0.0001)Multivariate analysis VEGFA rs2010963 [Exp(B): 2.77, P = 0.009] and ECOG performance status [Exp (B): 2.80, P = 0.004] were independent predictors of OS. The combination of these two parameters further stratified patients into three groups with progressively different OS (P < 0.0001)VEGFA rs2010963 was the only independent predictor of PFS (P = 0.005).CCL4 rs1634517-CC and CCL3 rs1130371-GG were associated with longer PFS in the evaluation set (2.5 mo vs 2.0 mo, HR: 1.54, P = 0.043; 2.5 mo vs 2.0 mo, HR: 1.48, P = 0.064) and longer PFS (2.3 mo vs 1.8 mo, HR:1.74, P < 0.001; 2.3 mo vs 1.8 mo, HR: 1.66, P = 0.002) and OS (7.9 mo vs 4.4 mo, HR: 1.65,P = 0.004; 7.9 mo vs 4.4 mo, HR: 1.65, P = 0.004) in the validation set. These associations were confirmed by multivariate analysis In the evaluation set, the CCL5 rs2280789-GG genotype vs the A allele (12.9 mo vs 7.9 mo, HR: 0.45, p = 0.032) and the rs3817655-TT genotype respect to A allele (12.9 mo vs 7.9 mo, HR: 0.50, P = 0.055) was associated with longer OS by multivariate analysis. The analysis in the validation set was not possible because of the low SNP frequencies In the evaluation set, the CCL5 rs2280789-GG genotype was associated with higher incidence of grade ≥ 3 HFRS compared to the A allele (53% vs 27%, P = 0.078), and similarly, the CCL5 rs3817655-TT genotype vs the A allele (56% vs 26%, P = 0.026). The same variants in addition to the CCL5 rs1799988 were also associated with different distribution by genotype of the incidence of grade ≥ 3 hypertension In the validation set, the CCL5 rs2280789-G allele was associated with inferior incidence of grade ≥ 3 diarrhea vs AA genotype (0% vs 10%, p = 0.034)and ΚLF13 rs2241779-A allele with inferior incidence of grade ≥ 3 rash respect to CC genotype (10% vs 30%, P = 0.010). CCL3 rs1130371 and CCL4 rs1634517 were associated with different distributions by genotype of the incidence of grade ≥ 3 AST/ALT Clinical endpoint PΚ Toxicity PFS OS DCR OSToxicity Therapy Regorafenib Toxicity Regorafenib Regorafenib Regorafenib Regorafenib PFS Study population Advanced solid cancer (n= 37; no HCC)(Japanese)Advanced solid cancer (n= 28; no HCC)(Japanese)refractory mCRC(n = 93)1(whites)mCRC (n =59)(whites)mCRC (n =79 Japanese patientsevaluation set;n = 150 Italian patientsvalidation set)Pharmacogenetic panel 3 SNPs in SLCO1B1,ABCG2 3 SNPs in SLCO1B1,ABCG2 Sequencing ofCYP34,UGT1A9 17 SNPs in VEGFA,VEGFC, FLT1 (VEGFR1),ΚDR (VEGFR2), FLT4(VEGF3)9 SNPs in CCL3, CCL4,CCL5, CCR5, PRΚCD,ΚLF13, and HIF1A 1Focused on 3 patients presented severe toxic hepatitis. ALT: Alanine aminotransferase; AST: Aspartate aminotransferase; DCR: Disease control rate; HCC: Hepatocellular carcinoma; HFSR: Hand-foot skin reaction; mo., months;OR: Odds ratio; OS: Overall survival; PFS: Progression-free survival; PΚ: Pharmacokinetic; SNP: Single nucleotide polymorphism.
Markers of pharmacokinetics/toxicity:(1) Sofarenib metabolism: The metabolism[29,30]of sorafenib is well-established and occurs mainly in the liver through two pathways:Phase I oxidation mediated by cytochrome P450 3A4 (CYP3A4), and phase II conjugation mediated by UDP glucuronosyltransferase 1A9 (UGT1A9) (Figure 2). In people, specifically, the glucuronidation contributes to about 15% of the clearance of sorafenib while the oxidation accounts for only 5%. Eight metabolites of sorafenib have been identified (M1-M8). The most abundant in the plasma is sorafenib N-oxide(M2), which is produced by CYP3A4 and exhibits an in vitro potency similar to the parental drug. M2 together with the sorafenib derivatives M4, obtained by demethylation, and M5, an oxidative metabolite, inhibit VEGFR and PDGFR signaling and members of the MAPΚ pathway. Given the key role of CYP3A4 and UGT1A9 in sorafenib metabolism, inducers or inhibitors of these enzymes, such as some foods and co-administered drugs (e.g., carbamazepine, dexamethasone, phenobarbital,phenytoin, rifampin, rifabutin, St. John’s wort), could modify bioavailability of the agent. Moreover, even if sorafenib is not a substrate for the cytochrome isoforms CYP2B6, CYP2C8, and CYP2C9 and the UDP glucuronosyltransferase UGT1A1, the biological agent in vivo inhibits activity of these enzymes with potential pharmacological consequences and drug-interaction events. Membrane translocation of sorafenib and its metabolites, including the inactive sorafenib-glucuronide (SG)derivative, has been reported to be carried out by the coordinated activity of ATPbinding cassette (ABC) and solute carrier (SLC) transporters, not yet all identified[31](Figure 2). An enterohepatic recirculation of sorafenib has specifically been suggested[31]; according to this hypothesis, the drug glucuronide-conjugated SG is extensively extruded from the hepatocytes into the bile through a process mediated mainly by the multidrug resistance protein (MRP) 2 (encoded by AΒCC2). However,under physiological conditions, a considerable fraction of intracellular SG can also be secreted back into the blood by some sinusoidal transport mechanisms, including MRP3 (encoded by AΒCC3). From the circulation, downstream hepatocytes can efficiently take up SG again via the organic anion transporter family member 1B(OATP1B1 and OATP1B3, encoded by SLCO1Β1 and SLCO1Β3)-type carriers,resulting in only low SG concentrations reaching the general circulation. This secretion-and-reuptake loop may help prevent saturation of MRP2-mediated biliary SG secretion in hepatocytes located upstream within liver lobules, resulting in more efficient drug detoxification. Once secreted into the bile, SG enters the intestinal lumen, where it can be a substrate for bacterial β-glucuronidases that regenerate the parental drug sorafenib. This sorafenib can then undergo intestinal absorption, thus reentering the circulation. This ongoing enterohepatic recirculation of sorafenib has been inferred to contribute to the long-lasting sorafenib plasma levels observed in patients. In addition to these transporters, preclinical in vitro studies have identified other membrane carriers that might translocate sorafenib and its metabolites, such as the hepatic uptake pump organic cation transporter-1 (OCT1, encoded by SLC22A1)and the efflux transporters P-glycoprotein (p-gp or MDR1, encoded by AΒCΒ1) and breast cancer resistance protein (BCRP, encoded by AΒCG2)[30].
Functional polymorphic variants in genes encoding the phase I and II enzymes and ABC/SLC transporters involved in the sorafenib pathway have been described and could contribute to the inter-individual variability in the pharmacokinetics and toxicity profile observed in patients treated with sorafenib. Some studies have evaluated the role of genetic polymorphisms in predicting the bioavailability and toxicity of sorafenib administered to patients with HCC (Table 1). The most consistent data concern the predictive contribution of germ-line genetic variants in the oxidative and glucuronidative pathways on outcome with sorafenib.
(2) Oxidation pathway: Guo et al[32]recently focused on some CYP450 polymorphisms. In preclinical aflatoxin-induced HCC rat models, CYP3A4*1B (rs2740574;located in the 5’ untranslated region [5’UTR]) and CYP3A5*3 (rs776746; located in the intron 3) variants were associated with the lowest and highest sorafenib plasma concentrations, respectively. This difference in drug disposition was consistent with a different toxicity risk; CYP3A5*3-carrier rats had the most severe liver (measured as a change in alanine aminotransferase [ALT] and AST blood concentration [IU/L] over time) and renal (measured as a change in blood urea nitrogen [nmol/L] and creatinin[umol/L] blood concentration [IU/L] over time) injury, whereas CYP3A4*1-carrier rats had the mildest toxicity outcome. This author group analyzed other CYP family genetic variants in the same study, using additional engineered rat models. Carriers of CYP2C19*2 (rs4244285; Pro227Pro) or CYP2D6*10 (rs1065852, Pro34Ser) had sorafenib plasma levels and associated liver/renal toxicity that were intermediate between those of rats carrying CYP3A5*3 or CYP3A4*1 genetic variants[32]. This preclinical observation on rat models was confirmed in a small group of Chinese patients with advanced hepatitis B and C viral-associated HCC treated with sorafenib. In these patients, the CYP3A5*3 polymorphism was associated with rapid worsening of hepatic damage, but CYP3A4*1 carriers showed only a small effect. The findings therefore suggested that the CYP3A5*3 variant that determines decreased CYP3A5 enzymatic activity[33]could influence hepatic and renal exposure to sorafenib, with severe associated damage.
(3) Glucuronidation pathway: Other investigations generated positive preliminary data on the predictive contribution of genetic variants in the glucuronidation pathway on sorafenib treatment outcome[34-36]. A study in a cohort of white patients with advanced solid cancer, including HCC, identified the rs17868320 variant in the promoter region of the UGT1A9 gene as a predictive factor for grade ≥ 2 diarrhea occurrence. Carriers of the polymorphic rs17868320-T allele were exposed to a higher toxicity risk, without any impact on systemic drug exposure[34]. To explain this result,the authors suggested that the increased intestinal expression of UGT1A9, linked to the rs17868320 polymorphism[37,38], could cause a higher glucuronidation rate of the sorafenib metabolite M6. M6 is the major sorafenib derivative found in the feces, and when it is converted by UGT1A9 to the glucuronidated form, it exerts a damaging action on enterocytes, provoking diarrhea. The discovery of novel predictive factors of sorafenib-induced diarrhea is of particular interest, not only for the effect on patient quality of life but also for a potential interference with oral absorption of the drug,leading to decreased anti-tumor efficacy.
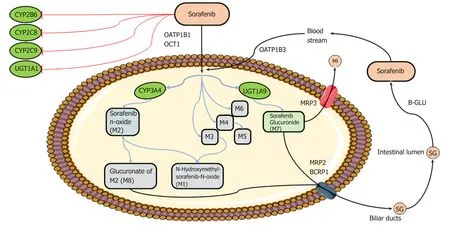
Figure 2 Schematic overview of sorafenib metabolism. Briefly, after oral administration, sorafenib enters hepatocytes by anion transporter family member(OATP1B, encoded by SLCO1B)-type carriers and cation transporter-1 (OCT1, encoded by SLC22A1). Within the hepatocytes, sorafenib undergoes phase I cytochrome P450 3A4 (CYP3A4)- and phase II UDP glucuronosyltransferase 1A9 (UGT1A9)-mediated metabolism to form M1-8 metabolites and sorafenib glucuronide (SG). After conjugation, SG is extensively secreted into the bile by a process that is mainly mediated by multidrug resistance protein (MRP) 2 (encoded by ABCC2) and breast cancer resistance protein BCRP (encoded by ABCG2) and into the bloodstream by MRP3 (encoded by ABCC3). A fraction of SG enters the intestinal lumen, where it could be a substrate for bacterial β-glucuronidases (B-GLU) that regenerate the parental drug sorafenib, which reenters the systemic circulation through the OATP1B3 carrier. CYP2B6, CYP2C8, CYP2C9, and UGT1A1 may interfere with sorafenib metabolism, being inhibited by sorafenib (see text for details). Image created with Servier Medical Art (https://smart.servier.com/).
Results of another study involving Κorean patients with intermediate-stage HCC receiving sorafenib in combination with TACE suggested that the genetic polymorphisms in UGT1A9 could also influence the development of HFSR[36]. This common side-effect shows an ethnicity-specific incidence (i.e., higher incidence in Asian trials compared with Western trials) and can affect treatment efficacy, causing dose reduction or treatment discontinuation[36]. Particularly, the A allele of the intronic variant UGT1A9 rs7574296, whose functional impact is not yet known[39], is associated with increased HFSR risk. This preliminary result is of great clinical interest because early detection of patients at risk for HFSR would allow for continuation of lifeprolonging therapy with minimal morbidity. Positive data also have been reported for an additional UGT1A isoform, UGT1A1, and its promoter polymorphism UGT1A1*28(rs8175347). A study involving predominantly white patients with advanced solid tumor, mostly HCC, identified the UGT1A1*28 variant as a clinically significant predictive factor in hyperbilirubinemia risk during the first 2 months of sorafenib treatment and consequently of treatment interruption risk[35]. The UGT1A1*28 allele also showed a trend to increased risk of developing any kind of toxicity of grade 3 or higher. These results are consistent with a previous case report reporting severe unconjugated hyperbilirubinemia in a sorafenib-treated patient carrying one UGT1A1*28 polymorphic allele[40]. This genetic variant is associated with a remarkable reduction in bilirubin glucuronidation activity of the UGT1A1 enzyme, leading to significantly increased bilirubin concentrations[14], and also sorafenib inhibits the same enzyme UGT1A1[41]. Thus, use of sorafenib in patients who are homozygous for UGT1A1*28 could lead to acute hyperbilirubinemia and a related risk of treatment interruption. Clinicians might need to be aware of their patient’s UGT1A1*28 status to adequately consider sorafenib therapy in cases of hereditary genetic predisposition to hyperbilirubinemia development (e.g., patients with Gilbert’s syndrome)[40].
(4) Transporter mechanism: Genetic variants in the sorafenib transporter mechanism also appear to influence drug availability and toxicity risk, although data are quite preliminary. Particularly, some exploratory studies involving white patients with advanced solid cancer, including HCC, and receiving sorafenib reported an association of some functionally relevant genetic variants in AΒCG2, AΒCΒ1, and SLCO1Β1 genes with sorafenib pharmacokinetics and pharmacodynamics[34,35,42]. The TT genotype for the intronic AΒCG2 rs2622604 polymorphism was associated with decreased protein expression[43], and patients treated with sorafenib and carrying the TT genotype showed a tendency toward higher drug exposure at the plasma level.This tendency was not, however, confirmed in the multivariate analysis probably because of the small population[34]. Tandia and colleagues also reported an impact of AΒCG2 variants on sorafenib bioavailability[42]. In their analysis, the heterozygous genotypes of AΒCG2 rs2231137 (Val12Met), AΒCG2 rs2231142 (Lys141Gln), and AΒCΒ1 rs2032582 (lle1145Ile) polymorphisms were associated with lower drug plasma levels in comparison to the wild-type genotype carriers. Another group focused instead on sorafenib-related toxicity and reported significant differences in toxicity incidence according to two SLCO1Β1 polymorphisms that alter the transport activity of OATP1B1 in a substrate-specific manner[44]: SLCO1Β1 rs2306283 (*1b,Asn130Asp) and SLCO1Β1-rs4149056 (*5, Val174Ala). Patients carrying at least one SLCO1Β1*1b (rs2306283-G) allele showed a reduced incidence of diarrhea and increased risk for hyperbilirubinemia; patients with the SLCO1Β1*5 (rs4149056-C)allele were more likely to develop thrombocytopenia, but only in a univariate and not in a multivariate model[35].
For background, we note that variants in MRP2- and OCT1-encoding genes also have been suggested to modulate sorafenib bioavailability and related adverse reactions, although mostly in other cancers. Studies performed in cancer settings other than HCC reported a significant involvement in the modulation of sorafenib plasma level and toxicity risk (e.g., erythema) for the promoter variant rs717620 in the AΒCC2 gene (encoding MRP2)[45,46]. Particularly, a preliminary investigation, which involved mainly white patients (n=120) with solid cancer receiving sorafenib, suggested that the AΒCC2 rs717620-TT polymorphic genotype was associated with the lowest sorafenib plasma concentration (i.e., AUC, area under the curve) compared with CT or CC genotype; interestingly this polymorphism seemed to modify AUC phenotype only in patients with UGT1A1*28/*28 status. Another study, including 55 Japanese patients with advanced renal cell carcinoma treated with sorafenib, indicated that the AΒCC2 rs717620-CC genotype was associated with significantly increased risk of developing grade 3 or higher HFSR respect to CT genotype (35 vs. 0%, P=0.032). For what concerns OCT1 genetic variants, an ex vivo investigation of HCC tumor samples demonstrated that two novel exonic polymorphisms in the SLC22A1 (gene encoding OCT1) (i.e., Arg61Ser fs*10 and Cys88Ala fs*16) were associated with decreased expression of the OCT1 transporter and dramatically affected the ability of sorafenib to reach active intracellular concentrations[47].
Markers of response:(1) Mechanism of action: Sorafenib exerts its pharmacological effect through inhibition of cell surface and downstream intracellular kinases involved in several tumor cell signaling pathways, including proliferation,angiogenesis, and apoptosis. Therefore, data from in vitro analysis and animal models have demonstrated that sorafenib exerts its anticancer activity by repressing proliferation of HCC cells and tumor growth, inducing HCC cell apoptosis, and reducing tumor angiogenesis and related pathways (e.g., inflammation)[29,30]. In addition to kinase inhibition, other mechanisms implicated in the activity of sorafenib include MAPΚ-independent apoptosis induction and immunomodulatory effects.Thus, primary and acquired resistance to sorafenib represent complex and multifaceted phenomena for which underlying mechanisms are not completely defined. At present, few pharmacogenetic studies have investigated the role of inherited genetic variability in determining the response to sorafenib (Table 2).
(2) VEGF-dependent pathways: The retrospective multicenter study ALICE1(Angiogenesis Liver CancEr) evaluated a panel of functionally relevant polymorphisms in genes encoding VEGF and its receptor VEGFR for their role in clinical outcomes among white patients with advanced or intermediate-stage HCC receiving sorafenib[48]. On univariate analysis, the rs25648-C, rs833061-T, rs699947-C, and rs2010963-C alleles in VEGFA, rs4604006-T allele in VEGFC, rs664393-G allele in FLT1(encoding the receptor VEGFR1), and rs2071559-C and rs2305948-C alleles in KDR(encoding the receptor VEGFR2) emerged as potential predictive markers of longer PFS and OS. At the multivariate level, VEGFA rs2010963-C and VEGFC rs4604006-T alleles, together with BCLC stage, were confirmed as the only independent prognostic factors predicting outcome in terms of PFS and OS. Moreover, the combination of VEGFA rs2010963 and VEGFC rs4604006 markers further improved patient stratification according to recurrence risk and survival probability. Patients expressing both favorable alleles showed longer PFS and OS compared to those expressing only one or none. The same favorable alleles were also significantly associated with a better objective response. The significant impact of VEGFA rs2010963 and VEGFC rs4604006 genetic variants, alone and in combination, on PFS and OS was also confirmed in the subsequent multicenter study ALICE-2[49].Collectively, these findings suggest an impact of polymorphisms that might influence the level of circulating VEGF, such as rs2010963, located in the 5’UTR region of the VEGFA gene, and rs4604006, located in one of the intronic sequences of the VEGFC gene. The result would be a crucial effect on a drug such as sorafenib that targets this pathway. Another study also confirmed the key involvement of the angiogenesis process in modulating sorafenib treatment. Results from this Chinese cohort with advanced HCC suggested positive results with polymorphisms in KDR encoding the receptor VEGR2, whose dysfunction is correlated with decreased antiapoptotic effects of VEGF among other vascular alterations[50]. Particularly, the AA genotype of the rs1870377 variant was associated with longer time to progression and with OS as well as with better objective response. The T allele of the rs2071559 variant was associated with longer OS. Both polymorphisms were reported to affect VEGFR2 functionality and/or expression level, thus potentially interfering with sorafenib’s mechanism of action[50]. The rs1870377 allele is a missense variant (Gln472His) located in the fifth NH2-terminal Ig-like domains within the extracellular region, which are important for ligand binding. Rs1870377, which is linked to a significant decrease in VEGF binding efficiency to VEGFR2, causes an altered protein phosphorylation pattern. Rs2071559 is a promoter variant that alters the binding affinity of this regulatory region for the transcriptional factor E2F, leading to decreased expression of the VEGF receptor. The same group reported preliminary data for another functionally relevant missense polymorphism, rs2305948 (Val297Ile), located in the third NH2-terminal Ig-like domains of the receptor. This variant was associated with differences in progression risk, with longer time to progression for the AA genotype, but only in the univariate and not in the multivariate model.
(3) Other pathways: Pharmacogenetic interest also has focused on different genetic targets in VEGF-dependent pathways. In particular, the Italian multicenter ePHAS study[51]focused on polymorphisms in the endothelial nitric oxide synthase (eNOS)gene, given the direct correlation between activation of the VEGF signaling pathway and stimulation of the vasodilator nitric oxide. This study, including training and validation populations of white patients with HCC undergoing sorafenib treatment,found in both cohorts a significant association of lower PSF and OS with a specific eNOS haplotype (i.e., HT1:T-4b), derived by the combination of a rs2070744 T-to-C substitution in the 5’UTR region and the intronic VNTR 27bp 4a/4b polymorphism(i.e., “4a” the allele with 4 repeats and “4b” the allele with 5 repeats). The rs2070744 variant was suggested to coordinate with the VNTR 27bp 4a/4b variant and directly affect gene transcription efficiency, resulting in altered eNOS expression levels that could in turn affect activation of VEGF signaling, and eventually sorafenib cytotoxicity. Particularly, the rs2070744-T and VNTR 27bp 4b alleles seemed to be associated with higher eNOS protein levels and activity, and consequently with increased basal NO production that could contribute to the sorafenib resistance. On the other hand, more recent preliminary results of another multicenter study, the ALICE-2[49], have highlighted a predictive role of polymorphisms in the gene encoding hypoxia-inducible factor α subunit (HIF1α) on sorafenib efficacy. HIF1α stabilization in hypoxic conditions upregulates VEGF expression by binding the VEGFA promoter,increasing angiogenesis. For this reason, HIF1α represents another player in the VEGF-dependent pathway that could be involved in sorafenib efficacy. Moreover,overexpression of HIF-1α in HCC is associated with tumor angiogenesis, invasion,metastasis, treatment resistance, and poor prognosis. The ALICE-2 study, which involved white patients with HCC treated with sorafenib, showed that HIF1A rs1951795, rs10873142, and rs12434438 variants contribute to discriminating patients according to different progression and survival probabilities. Multivariate analysis confirmed the predictive role only for the HIF1A rs124344308 polymorphism with the GG genotype, associating it with poorer PFS and OS independently from VEGF markers (i.e., VEGFA rs2010963; VEGFC rs4604006). An additional clinical study[52]with a similar patient cohort generated positive data for genetic markers in another key angiogenic factor, Ang-2. By binding to its receptor Tie2, Ang-2 cooperates with the VEGF pathway in regulating angiogenesis and maintaining normal physiological vascular functions. In cancer, this protein is suggested to contribute to determining tumor aggressiveness and metastatic phenotype. In addition, a high baseline level of Ang-2 correlates with shorter OS in patients with advanced HCC without affecting clinical response to sorafenib[53]. A preliminary study by Marisi et al[52]explored for the first time the role of an Ang-2 genetic variant in sorafenib therapy outcome. These authors found that in particular, the GG genotype of the synonymous polymorphism rs55633437 (Thr238Thr) was associated with significantly longer PFS and OS compared to other genotypes.
Regorafenib
Regorafenib (STIVARGA®) is an oral small molecule inhibitor with an almost identical structure to sorafenib with which it shares most of the pharmacokinetic and pharmacodynamic properties[54]. Regorafenib, similarly to sorafenib, blocks multiple membrane-bound and intracellular kinases involved in normal cellular functions and pathologic processes such as tumor angiogenesis (VEGFR1, -2, -3, TIE2), oncogenesis(ΚIT, RET, RAF-1, BRAF), and modulation of the tumor microenvironment (PDGFR,FGFR). However, the small but significant difference in the chemical structure confers on regorafenib a stronger inhibition power of the targeted angiogenic and oncogenic kinases than sorafenib, resulting in higher pharmacological potency[54]. The liver metabolism of regorafenib, even if less well-characterized, is comparable with that of sorafenib and occurs through an oxidative process mediated by CYP3A4 and glucuronidation mediated by UGT1A9[54]. Two major and six minor metabolites of regorafenib have been identified in human plasma. The main circulating metabolites are M2 (N-oxide) and M5 (N-oxide and N-desmethyl), which show similar steadystate plasma concentrations and efficacy compared to the parental drug, as studied in in vitro and in vivo models[54-56]. Moreover, regorafenib and its metabolites M2 and M5 are suggested substrates of some ABC/SLC membrane transporters, such as MDR1,BCRP, MRP2, and OATP1B1, and thought to undergo enterohepatic recycling similar to that of sorafenib[54-56]. Regorafenib and its major metabolites are also reported to inhibit a number of cytochromes (CYP2C8, CYP2C9, CYP2B6, CYP3A4, CYP2D6),UGT1A enzymes (UGT1A9, UGT1A1), and transporters (BCRP) and induce others(CYP1A2, CYP2B6, CYP2C19, CYP3A4) with potential alteration in the exposure of coadministered drugs[55-58].
Since the recent introduction of regorafenib as a second-line treatment for HCC, no pharmacogenetic data have been published regarding potential genetic markers that could predict the risk of severe toxicity and response to the targeted drug in patients with liver cancer. However, given the similar metabolism and mechanism of action between regorafenib and sorafenib, the same genes and related variants suggested to modulate sorafenib therapy may also influence regorafenib. In support of this hypothesis are preliminary results from recent studies performed in other cancer settings, where regorafenib has been used for a long time. Details regarding the pharmacogenetic panel analyzed, the study population (e.g., disease, sample size,ethnicity) and therapy (e.g., dose and schedule) characteristics, the clinical end-points evaluated along with the main findings (e.g., statistical results) of the studies are shown in Table 3.
Markers of pharmacokinetics/toxicity:Regarding potential markers of regorafenib toxicity, preliminary positive data have been generated for variants in genes encoding the metabolic enzymes UGT1A9, BCRP, and OATP1B1. A descriptive study[57]assessed CYP3A4 and UGT1A9 genetic variability by sequencing the germline DNA of three patients with metastatic colorectal cancer (mCRC) experiencing severe toxic hepatitis after sorafenib treatment and reported that two patients were heterozygous for the UGT1A9*22 (rs3832043) polymorphism. This variant consisted of a single base insertion of thymidine in the promoter region and it is likely to increase gene expression and enzymatic function[59]. The high-activity UGT1A9*22 allele probably affects hepatic metabolism of regorafenib, setting the stage for hepatotoxicity. This finding warrants strict liver monitoring during regorafenib treatment for patients with unfavorable UGT1A9 genotypes. However, further investigations are needed to explore the exact mechanism by which an altered activity of UGT1A9 could contribute to the occurrence of hepatotoxicity. Another preliminary investigation of a small cohort of Japanese patients with solid cancer and receiving regorafenib[60]showed that the presence of the SLCO1Β1*1b (rs2306283-G) allele protected against the development of grade ≥ 2 hepatic injury and anemia, two of the most important regorafenib-related adverse drug reactions. The authors speculated that these associations could arise from a change in the pharmacokinetic profile of the biological agent, resulting from an inherited alteration in transporter activity of OATP1B1, as determined by the functional *1b variant haplotype[44]. The same work also showed that the loss-of-function rs2231142-A allele of the AΒCG2 gene correlated with inferior blood platelet counts (Plt) without an effect on risk for treatment-related grade ≥ 2 adverse reactions. A subsequent study from the same group[61], monitoring a small cohort of Japanese patients with solid cancer for 28 days after regorafenib administration, showed a tendency, although not significant, to a higher drug concentration-to-dose ratio for the SLCO1Β1*5 (rs4149056-C) allele and for SLCO1Β1*1b (rs2306283-G/rs4149056-T) non-carriers. However, further investigations are required to confirm this association and to understand the biological mechanism underlying the observed genotype/phenotype correlation. Of interest, the results of the same study also included a strong association between serum regorafenib concentrations and total bilirubin levels, which could be used as a potential marker for estimating regorafenib pharmacokinetics. In fact, liver cells take up unconjugated bilirubin through OATP1B1, and in the hepatic cell, it is conjugated to glucuronic acid by UGT1A1. Considering that serum bilirubin is suggested to increase because of competitive inhibition via OATP1B1, bilirubin plasma level could be considered a surrogate marker of drug exposure. However, further analyses are needed to clarify the exact mechanism of competition between regorafenib and bilirubin with respect to OATP1B1.
Beside polymorphisms in metabolic enzymes encoding genes, genetic variants affecting the VEGFA-related pathway are also hypothesized to contribute to interindividual differences in toxicity risk. Particularly, a recent study[62], including an evaluation (Japanese mCRC patients) and validation (Italian mCRC patients) cohort,reported significant differences in toxicity incidence according to genetic variants in the C-C motif chemokine ligand 5/C-C motif chemokine receptor 5 (CCL5/CCR5)pathway. This pathway modulate VEGFA production via endothelial progenitor cell migration. The investigation showed that in the evaluation set, the CCL5 rs2280789-GG and rs3817655-TT genotypes were associated with higher incidence of grade ≥ 3 HFRS. The replication of these associations according to the recessive model in the validation set was not possible because of the low frequency of the homozygote genotype. With respect to the risk for HFRS, the observed differences in the frequency distribution of the rs2280789 and rs3817655 variants between the Japanese and Italian cohorts could also explain the different incidence of severe HFRS by ethnicity noted in clinical practice[36]. An exploratory analysis of the other toxicity types was also performed and highlighted that, in the evaluation set, the CCL5 rs2280789, rs3817655,and rs1799988 variants could have a predictive effect on risk for grade ≥ 3 hypertension. In the validation set, the CCL5 rs2280789 variant emerged as a predictive marker of grade ≥ 3 diarrhea while the CCL4 rs1634517 and CCL5 rs1130371 markers were differently distributed in genotype frequencies relative to incidence of grade ≥ 3 AST/ALT variation. Another marker of the CCL5/CCR5 pathway, the KLF13 rs2241779, seemed to influence risk for grade ≥ 3 rash. Although these findings should be considered exploratory, they suggest a promising candidate targets for future pharmacogenetic studies aimed at discovering novel predictive markers to improve the management of regorafenib-associated toxicity (e.g., personalized dosing and other strategies to support patient care).
Markers of response:Other studies have focused on the potential role of polymorphisms in the VEGF/VEGFR cascade and related mechanisms in modulating the response to regorafenib treatment. The work of Giampieri and colleagues[63],involving a small cohort of white patients with mCRC, reported that the VEGFA rs2010963 variant is an independent predictive marker of regorafenib efficacy in terms of disease control rate, PFS, and OS, with the CC genotype associated with a better outcome. The integration of patient Eastern Cooperative Oncology Group (ECOG)performance status with the VEGFA rs2010963 genotype improved stratification by survival rate. On univariate analysis, other markers, such as VEGFR2 rs1870377,VEGFR3 rs307805, and VEGFR1 rs664393, were suggested to contribute to determining regorafenib outcome. In particular, the VEGFA rs2010963 variant, located in the 5’UTR of the gene, has a potential effect on VEGFA expression and tumor angiogenesis. The observed association is consistent with the results of other studies reporting significant involvement of VEGFA rs2010963 in influencing other biological agents targeting the VEGF/VEGFR cascade, such as sorafenib and bevacizumab[48,64].
Another recent investigation[62]evaluated the role on regorafenib therapy outcome of a panel of variants from the CCL5/CCR5 pathway that is involved in the modulation of VEGFA production. The study comprised 79 Japanese patients with mCRC as the evaluation cohort and 150 Italian patients with mCRC as the validation cohort. The results showed that in the evaluation set, the CCL5 rs2280789-GG and rs3817655-TT genotypes were associated with longer OS. The replication of these associations according to the recessive model in the validation set was not possible because of the low frequency of the homozygote genotype. Functional analyses have demonstrated that the G allele of the rs2280789 polymorphism, located in the promoter region of CCL5, negatively affects transcriptional activity of RANTES,resulting in a lower serum level of CCL5 and VEGFA[62]. A similar phenotypic effect on CCL5 and VEGFA expression level was also suggested for the T allele of the intronic CCL5 rs3817655 variant[62]. These functional data could help explain the clinical impact on regorafenib outcome observed for the CCL5 rs2280789 and rs3817655 markers. Of interest, the same study generated positive data for polymorphisms in genes encoding other CCR5 ligands, such as CCL4 (rs1634517,intronic variation) and CCL3 (rs1130371, synonymous variation, Pro60Pro) that were associated with PFS and OS in both evaluation and validation cohorts. These variants also displayed similar allelic distribution between the two ethnic groups, unlike CCL5. From a functional point of view, the CCL4 rs1634517-C and CCL3 rs1130371-G alleles, associated with longer PSF and OS, seemed to correlate with higher CCL5 level without any impact on VEGFA level[62].
Taken together, these data highlighted the importance of the VEGF/VEGFR cascade and related pathway (i.e., CCL5/CCR5) in modulating the effectiveness of regorafenib therapy. Polymorphisms in gene encoding the several members of these pathways should be the target of future pharmacogenetic studies aimed at optimizing regorafenib treatment outcomes.
Other approved drugs
Cabozantinib (XL-184, COMETRIQ®) is an oral tyrosine kinase inhibitor that can block multiple oncogenic and angiogenic pathways implicated in tumor progression, worse prognosis, and metastasis, such as PDGFR, HGFR, VEGFR2, AXL, RET, ΚIT, and FLT3[65-69]. Following oral administration, the median time to peak plasma concentrations (Tmax) of cabozantinib ranged from 2 to 5 hours post-dose. This drug undergoes hepatic metabolism by CYP3A4 and, to a minor extent, by CYP2C9[66]. In addition, the major metabolites of cabozantinib identified in human plasma, after a single dose oral intake (140 mg), are EXEL-1646 (M9), obtained from M16 sulfation;EXEL-5162 (M19), obtained from the oxidation at the nitrogen of the quinolone portion; EXEL-5366 (M7), derived from the hydrolysis at the amide bond; and EXEL-1644 (M2a), the M7 sulfate conjugate[66,70]. Considering excretion, cabozantinib is eliminated mostly by the feces (54%) and urine (27%)[66]. Between 2012 and 2013, the FDA and the European Medicines Agency initially approved cabozantinib as a treatment for patients with medullary thyroid cancer. In 2016, the drug received a new indication as a treatment for patients with advanced renal cell carcinoma following one prior anti-angiogenic therapy[71-73]. Recently, several clinical trials have demonstrated that cabozantinib exhibits encouraging clinical activity in multiple human cancers, including HCC, with manageable side-effects[25,74-77]. Based on this evidence, cabozantinib represents an efficient alternative in the management of sorafenib-resistant HCC. In 2018, it received FDA approval for HCC treatment[76,78].
Lenvatinib (E7080 or LENVIMA®) is an orally active multikinase inhibitor that selectively inhibits receptors related to pro-angiogenic and oncogenic pathways such as VEGFR1-3, FGFR 1-4, PDGFRα, and RET, and ΚIT proto-oncogenes[79-83]. After oral administration, lenvatinib is rapidly absorbed, and time to peak plasma concentration occurs from 1 to 4 hours postdose[84]. However, even if administration with food does not affect the extent of absorption, it can decrease the rate of absorption and delay median Tmax from 2 to 4 hours. Both in vitro plasma and in vivo serum proteinbinding assays demonstrated that lenvatinib protein binding ranges from 96.6 to 98.2%[84]. Lenvatinib is metabolized in liver microsomes mostly through CYP3A4 (>80%) and, to a minor extent, by aldehyde oxidase and acts as a substrate for ABC transporters, encoded by the ABCB1 and ABCG2 genes, such as BCRP and Pglycoprotein[84-86]. Regarding excretion, the percentage of unchanged lenvatinib found in urine and feces is 2.5% of the administered dose, suggesting that lenvatinib is highly metabolized.
The principal metabolites of lenvatinib are derived from decyclopropylation (M1),demethylation (M2), N-oxidation (M3), and O-dearylation (M5)[84]. The formed metabolites are mainly excreted, approximately 64% via the biliary route in the feces,and 25% of the metabolites formed in the liver are released into the circulation and excreted via urine[84,87]. At first, lenvatinib was approved for the treatment of radioiodine-refractory differentiated thyroid cancer, as a single agent, and for the treatment of advanced renal cell carcinoma in combination with everolimus[79,88-90]. On August 2018, based on positive results of the REFLECT trial (NCT01761266), the FDA approved lenvatinib as a first-line treatment in patients with advanced and unresectable HCC[91,92].
To the best of our knowledge, no studies still have investigated the correlation between genetic polymorphisms and cabozantinib or lenvatinib treatment outcome for either toxicity or efficacy in HCC patients. However, a very recent study by Ozeki et al93on Japanese patients with thyroid cancer, demonstrated for the first time an impact of CYP3A4/5 and AΒC transporter genetic variants on lenvatinib pharmacokinetics[93]. Particularly, the CYP3A4*1G (rs2242480, intronic variation) and AΒCC2 rs717620 polymorphisms were suggested to have an effect on the steady-state mean plasma [i.e., mean dose-adjusted C0, (ng/mL/mg)] trough concentrations of lenvatinib. The mean dose-adjusted C0values of lenvatinib in patients with the CYP3A4*1/*1 genotype and AΒCC2 rs717620-T allele were significantly higher than those in patients with the CYP3A4*1G allele and AΒCC2 rs717620-CC genotype,respectively (effect size: 0.863, P = 0.018 and effect size: 0.605, P=0.036, respectively).Moreover, the dose-adjusted C0of lenvatinib in patients with both the CYP3A4*1/*1 genotype and AΒCC2 rs717620-T allele (median 6.70 ng/mL/mg) was about 1.5-fold higher than that in patients with both the CYP3A4*1G/*1G and ABCC2 rs717620-CC genotypes (median 4.42 ng/mL/mg; P = 0.007)[93]. These results demonstrated that functionally relevant genetic variants in proteins involved in the metabolism,translocation, and mechanism of action of cabozantinib or lenvatinib could be important determinants of therapy outcome and represent good candidates for future pharmacogenetic studies. With increasing therapeutic opportunities, the identification of markers that help clinicians choose the drug most suited to that patient becomes an urgent need. On this ground, Takeda et al. recently said that “approval of lenvatinib opened the new era of molecular targeting therapy for HCC. It requires the use of several molecular targeted agents appropriate for each HCC patient. To realize this personalized medicine, the establishment of genetic or transcriptional biomarkers needed to select the appropriate regimen is eagerly awaited’’[94].
PHARMACOGENETICS OF DRUGS UNDER INVESTIGATION
The genomic understanding of HCC and the development of molecularly targeted therapies represent a promising stepping-stone for increasing the number of effective drugs for HCC patients. In recent years, many new drugs have been tested or are still under investigation as an alternative to sorafenib or, most important, after sorafenib failure. However, even if the survival benefit improvement and adverse drug event reduction are still the main focus, the identification of predictors of good responders could allow application of these new drugs in personalized treatments for HCC[95-98].Furthermore, a deep understanding of the proteins involved in the metabolic pathway and mechanism of action of these novel molecularly targeted agents could suggest potential candidate targets (i.e., genes and polymorphisms) for future pharmacogenetic studies. Therefore, this paragraph is focused on drugs currently under investigation for HCC therapy by providing general information on their metabolism, pharmacokinetics, mechanisms of action and, where available,pharmacogenetics data.
Nivolumab
The presence of tumor-infiltrating lymphocytes expressing programmed cell death protein-1 (PD-1, encoded by PDCD1) in HCC lesions and their correlation with outcome paved the way for immunotherapeutic approaches for HCC treatment[98-101].The immune checkpoint inhibitor nivolumab (MDX-1106,OPDIVO®) is a fully human immunoglobulin (Ig) G4 (IgG4) monoclonal antibody. It binds the PD-1 receptor,expressed on activated T-cells, blocking interaction with its ligands PD-L1 and PD-L2 on tumor cells. This inhibition leads to downregulation of the T-cell-promoted tumor immune-escape mechanism, restoring the antitumor activity of T-cells[102,103].Nivolumab is intravenously administered and thus is completely bioavailable. After initiation of the infusion, its median time to peak concentration is 1-4 hours[104,105]. As stated on the drug label, no formal studies were conducted to characterize the specific nivolumab metabolic pathway. However, it is thought to be degraded into small peptides and aminoacids through canonical pathways, such as endogenous IgG, and not by CYPP450. Similarly, no studies have addressed the specific elimination route of nivolumab. The phase I/II CHECΚMATE-040 trial (NCT01658878) demonstrated the efficacy, safety, and tolerability of nivolumab in HCC treatment leading, on September 2017, to its accelerated FDA approval for the treatment of HCC in patients who previously have been treated with sorafenib[26,98]. At present, the multicenter phase III randomized controlled CHECΚMATE-459 trial (NCT02576509) is ongoing to determine if nivolumab or sorafenib is more effective as first-line treatment for advanced HCC. In term of pharmacogenetics, it has been demonstrated, in lung adenocarcinoma, non-small cell lung cancer (NSCLC) and squamous cell carcinoma,that PD-1/PD-L1 gene polymorphisms may alter the immune checkpoint functions and affect the clinical response to nivolumab[106,107]. Patients with the CC or CG PD-L1 genotypes (rs4143815) and the GG or GT PD-L1 genotypes (rs2282055) experience a significantly longer median PFS (2.6 months) with nivolumab treatment than patients with the GG and TT genotypes (2.1 and 1.8 months respectively)[106]. Furthermore,none of the patients obtained a treatment effect with the GG genotype of PD-L1 rs4143815 and the TT genotype of rs2282055. In addition, it has been demonstrated that rs2297136, rs4143815, and rs17718883 polymorphisms of the PD-L1 gene are associated with HCC risk and prognosis[107,108]. Even if the functional and biological effect of PD-L1 genetic variants are still under investigation and debate, taking together, these results reinforce the role of these polymorphisms as possible prognostic markers for HCC development as well as markers of outcomes in nivolumab-treated patients[108-110].
Another study analyzed 322 nivolumab-treated patients with NSCLC and assessed the association between toxicities and polymorphisms in genes considered as contributors to PD-1-directed T-cell responses, such as the PD-1 gene (PDCD1),tyrosine-protein phosphatase non-receptor type 11 (PTPN11) and interferon gamma(IFNG). The TT genotype in the PDCD1 rs2227981 polymorphism was associated with less nivolumab toxicity. On the contrary, patients presenting one G allele in the PTPN11 rs2301756 polymorphism or who are homozygous CC for the IFNG rs2069705 polymorphism were at increased risk for developing any grade toxicity[111]. Further investigations are required to confirm these preliminary data and to test their validity also in the HCC setting.
Pembrolizumab
Pembrolizumab (lambrolizumab or MΚ-3475 or ΚEYTRUDA®) is a high-affinity humanized IgG4 monoclonal antibody that can bind with to the cell surface receptor PD-1, antagonizes receptor interaction with its known ligands PD-L1 and PD-L2, and allows the immune system to destroy cancer cells[98]. The antibody, intravenously administered, is immediately and completely bioavailable, does not bind to plasma proteins, and undergoes catabolism to small peptides and single aminoacids via general protein degradation routes[112]. In terms of clearance, a correlation has been demonstrated between clearance rate and increasing body weight, explaining the rationale for dosing on an mg/kg basis, whereas age, sex, race, and tumor burden have no clinically important effect on clearance. Furthermore, mild or moderate renal and hepatic impairments do not differ in clinically important way in clearance compared to patients with normal functions[112]. In 2016, Truong et al. published the first case report of a 75-year-old man with advanced HCC responsive to pembrolizumab, on a compassionate use basis, after failure of sorafenib therapy[113]. Since 2016, several observational and interventional phase I/II/III studies, such as the ΚEYNOTE-224 and the ΚEYNOTE-240 trials, continue investigating the safety and efficacy of pembrolizumab, alone or in combination with other drugs/procedures, in patients with advanced HCC who progressed on or were intolerant to first-line systemic therapies (e.g., NCT02940496, NCT02658019, NCT03062358, NCT03753659,NCT02702401)[83,114]. In 2016, considering a cohort of patients with metastatic melanoma treated with pembrolizumab or nivolumab, it has been demonstrated that 28% of responsive tumors were significantly enriched in non-synonymous singlenucleotide variations in disparate breast cancer type 2 susceptibility protein (BRCA2)domains. Specifically, one in the N-terminal nucleophosmin-interacting region(rs775903570, Val950Leu), one in the DNA polymerase eta-interacting domain(Ser1792Phe), four in the helical domain critical for Fanconi anemia group D2(FANCD2) interaction (His2361Tyr [rs786203493], Pro2505Ser, Ser2522Phe,His2537Tyr), and one between these two interacting domains (Glu2115Lys)[115]. The authors, according to the disposition of the highlighted loss-of-function mutations and known role of BRCA2 in DNA repair, suggested that enhanced responsiveness could arise from cellular stress resulting from defective DNA repair that leads to increased cell death and anti-tumor immunity[115,116].
Furthermore, Al-Samkari et al[117]recently published a case report of a 58-year-old woman with aggressive metastatic breast cancer who developed hemophagocytic lymphohistiocytosis (HLH) while undergoing experimental treatment with pembrolizumab, resulting in critical illness and multi-organ system failure. Nextgeneration sequencing revealed that she was heterozygous for germ-line perforin-1(PRF1) c.272C>T (rs35947132, p.Ala91Val). Several studies have demonstrated that PRF1 rs35947132 is aberrantly post-translationally processed and results in reduced perforin expression together with partial loss of lytic activity. The rs35947132 polymorphism is a genetic risk factor for the development of HLH in patients exposed to certain environmental triggers. Taking all these findings together, the authors postulated that in the presence of the PRF1 polymorphism, pembrolizumab treatment could ignite a dramatic adverse drug event such as HLH[117]. Once again, these interesting pharmacogenetic results stress the hypothesis that the presence of genetic variations could affect, in this case, pembrolizumab therapy outcome, giving the possibility to investigate and, so, to extend their spectrum of action to other oncological fields, such as HCC therapy.
Palbociclib and ribociclib
Palbociclib (PD-0332991, IBRANCE®) and ribociclib (LEE-011,ΚISQALI®) are oral,specific inhibitors of the cyclin-dependent kinases CDΚ4 and CDΚ6[118,119]. Through CDΚ inhibition, both drugs prevent the formation of the cyclin D-CDΚ4/6 complex and retinoblastoma protein phosphorylation. Accordingly, cells cannot switch from R to G1 phase and proceed through the cell cycle[120,121]. In addition to canonical CDΚ4/6 retinoblastoma signaling, palbociclib shows in vitro and in vivo antiHCC activity by inducing cell autophagy and apoptosis via a mechanism involving 5’ AMPactivated protein kinase activation and protein phosphatase 5 inhibition[122]. Palbociclib is slowly absorbed, with a median Tmax generally observed between 6 to 12 hours, while ribociclib is rapidly absorbed, with median Tmax ranging from 1 to 5 hours. Binding of palbociclib to human plasma proteins in vitro is approximately 85%, while binding of ribociclib is approximately 70%, with no concentration dependence in either case.Following oral administration, palbociclib and ribociclib undergo extensive hepatic metabolism mainly by CYP3A; palbociclib also is metabolized through the sulfotransferase enzyme SULT2A1[123,124]. The major primary metabolic pathways for palbociclib involve oxidation and sulfonation, with acylation and glucuronidation contributing as minor pathways. For ribociclib, the primary metabolic pathways involve oxidation (dealkylation, C and/or N-oxygenation, oxidation (-2H)) and combinations thereof. Phase II conjugates of ribociclib phase I metabolites involved Nacetylation, sulfation, cysteine conjugation, glycosylation, and glucuronidation.Palbociclib and ribociclib are the major circulating drug-derived entities in plasma(23% and 46%, respectively), and their clinical activity traces primarily to the parent drug, with negligible contribution from circulating metabolites. Both drugs are eliminated mostly (69%-74%) via the feces, but also (17%-23%) via the urine.Following encouraging results from clinical trials, palbociclib and ribociclib have been approved, between 2015 and 2017, by the FDA and European Medicines Agency for hormone receptor-positive, human epidermal growth factor receptor 2-negative(HR+/HER2-) advanced or metastatic breast cancer therapy in combination with an aromatase inhibitor, such letrozole, or with fulvestrant (FASLODEX®), a selective estrogen receptor degrader, in women with disease progression after endocrine therapy[125-134]. The effects of palbociclib and ribociclib as a treatment for other malignancies, including HCC, are of great clinical interest and under current investigation (NCT01356628, NCT02524119).
Tivantinib
Tivantinib (ARQ197) is a selective, orally available, non-ATP competitive c-MET inhibitor currently under clinical investigation in patients with cancer[135,136]. Indeed,upregulation of the c-MET pathway, including its only known ligand HGF, is found in multiple cancers, such as HCC, and is associated with poor prognosis and metastases[136-139]. Conversely, tivantinib also revealed an anti-proliferative activity that was not restricted to only c-MET-dependent cell lines[140]. In fact, several in vitro and in vivo studies have demonstrated that tivantinib can affect microtubule dynamics by disrupting mitotic spindles. It also can promote G2/M cell cycle arrest and apoptosis by inhibiting the anti-apoptotic molecules myeloid cell leukemia-1 and B-cell lymphoma-extra large and increasing Cyclin B1 expression[140-144]. Indeed, despite controversies regarding its mechanism of action, two phase II clinical trials(NCT01575522, NCT00988741) have demonstrated that tumors with high levels of MET present a high degree of response to tivantinib treatment[145,146]. In term of pharmacokinetics, tivantinib is metabolized by CYP2C19, CYP3A4/5, UGT1A9, and alcohol dehydrogenase isoform 4[147]. CYP2C19 shows catalytic activity for the formation of the hydroxylated metabolite (M5), whereas CYP3A4/5 catalyzes formation of M5 and its stereoisomer (M4). Moreover, CYP3A4/5 represents the major cytochrome isoform involved in the elimination of M4, M5, and the ketometabolite (M8), and together with UGT1A9, involved in the glucuronidation of M4 and M5[147]. Finally, the alcohol dehydrogenase isoform 4, through a sequential ketometabolite of M4 and M5 and through M8, leads to the formation of M6[147]. Between 2010 and 2014, two phase I trials (NCT01069757, NCT01656265) in Japanese patients with advanced solid tumors examined the safety, pharmacokinetics, and preliminary efficacy of tivantinib as a single agent to determine recommended phase II dose according to CYP2C19 polymorphisms[148,149]. Recently, two Phase III trials, the METIVHCC (NCT02029157) and the JET-HCC (NCT01755767), were conducted to determine if tivantinib is effective as a second-line treatment MET in patients with diagnostichigh HCC who have already been treated once with another systemic therapy. This trial also further evaluated the safety profile of the experimental drug in this population[12,150]. Unfortunately, no statistically significant differences between tivantinib and placebo in terms of OS or PFS were identified in either trial.
CONCLUSION AND FUTURE DIRECTIONS
Despite the advantage in patient survival resulting from the introduction of targeted agents in the therapeutic scenario of HCC, a significant inter-individual heterogeneity in therapy outcome persists and constitutes a crucial problem in HCC management.Moreover, with the increasing number of therapeutic options, selection of the most appropriate treatment for each patient is a great challenge. The identification of genetic markers that predict which patients will benefit from a specific intervention could significantly affect decision-making and therapy planning
Most of the pharmacogenetic studies published to date have focused on sorafenib,which has the longest track record in HCC treatment. However, these investigations,conducted for the most part in small cohorts, have generated only quite sparse,unreplicated data that do not permit drawing conclusions about their clinical validity.Moreover, comparison among studies is difficult because of the high heterogeneity in regard to the ethnic and clinical-demographic characteristics of the cohorts, study design (e.g., retrospective/prospective analyses, low statistical power, adoption of an internal data validation), panel of investigated genes and related variants, method of controlling for confounding and environmental factors, and parameters for measuring clinical outcomes. For these reasons, the pharmacogenetic data published to date should be considered only hypothesis-generating. These data could be useful,however, for indicating specific genes and related pathways as potential candidate predictors of sorafenib therapy, guiding future research efforts.
Regarding the discovery of potential markers of toxicity risk after sorafenib administration, CYP and UGT1A polymorphisms are associated with different risks for severe adverse events. Although these findings should be considered preliminary because of small sample sizes, lack of replication, and some negative results[151], they surely support the need for additional pharmacogenetic research efforts to deeply understand the real clinical utility of CYP and UGT1A markers. Other published data,although mainly hypothesis-generating, have pointed out the clinical contribution in determining the sorafenib-related toxicity risk of polymorphisms in AΒC/SLC membrane transporter genes. Some caveats are necessary, however. Given the complex enterohepatic recirculation in which sorafenib appears to be involved (see above)[31], it could be supposed that the evaluation of the combinatorial effect of multiple ABC/SLC carriers with respect to single gene/variant analysis is a more effective strategy for identifying inter-individual differences in the pharmacological profile of sorafenib. Moreover, based on the results of a pilot study, evaluating UGT1A1*28 carriers with two distinct phenotypes in relation to sorafenib exposure based on AΒCC2 rs717620 genotypes[46], the integration of inherited variability in multiple metabolic processes, such as phase I and II metabolism and membrane translocation, could further improve prediction of therapy outcome. Moreover, the investigation of other pathways with closer links to the drug mechanism of action,such as angiogenesis (e.g., VEGFA) and inflammation (e.g., tumor necrosis factor-α),could be useful for discovering additional novel genetic markers that could contribute to stratifying patients based on individual toxicity risk[36].
Concerning potential genetic determinants of sorafenib efficacy, recent studies have highlighted the potential utility of inherited variability in the VEGF/VEGFR cascade and related pathways to identify patients who could benefit from sorafenib administration. This information can help clinicians in the selection of the most appropriate treatment and improve clinical outcome. For example, patients with a favorable genetic background could be administered sorafenib as soon as clinically indicated, instead of delaying it with other therapy; patients with an unfavorable background could be preferably included in clinical trials exploring new or upcoming treatment options. At present, the multicenter prospective INNOVATE study is ongoing to validate the contribution of polymorphisms in genes encoding VEGF,eNOS, Ang-2, and HIF-1α in determining clinical outcomes in patients with advanced HCC receiving sorafenib (NCT02786342)[152].
Only a few pharmacogenetic data, obtained for the most part in a non-HCC tumor setting, have been published regarding the recently approved regorafenib. Globally,these data, are only of hypothesis-generating value, but they have indicated potential candidate genes related to the regorafenib metabolism (e.g., ABC/SLC transporters,UGT1As) and mechanism of action (e.g., VEGFA and its receptors; the CCL5/CCR5 pathway). Their predictive power for therapy outcome could be useful to investigate in the HCC setting. Moreover, the similar pharmacological proprieties of regorafenib and sorafenib suggest that the genetic determinants of therapy outcome described for sorafenib could apply for regorafenib treatment and should be further investigated.
For the more recently developed multikinase kinase inhibitors (e.g., lenvatinib,cabozantinib), immune checkpoint inhibitors (e.g., nivolumab, pembrolizumab), and selective CDΚ4/6inhibitors (e.g., palbociclib, ribociclib), no pharmacogenetic markers have been identified in the HCC setting. Research efforts should respond to this lack of information.
The discovery of biomarkers, subsequently validated in large prospective studies, is a compelling need because they are expected to allow for more accurate selection of patients with HCC who are potential candidates for a specific targeted agent. This stratification could mean the ability to limit treatment to potentially responding patients and sparing unnecessary toxicity to those who are unlikely to benefit.
杂志排行

World Journal of Gastroenterology的其它文章
- Improving cirrhosis care: The potential for telemedicine and mobile health technologies
- Lumen-apposing metal stents for malignant biliary obstruction: Is this the ultimate horizon of our experience?
- Consensus on management of hepatitis C virus infection in resource-limited Ukraine and Commonwealth of Independent States regions
- Immunotherapy in colorectal cancer: Available clinical evidence,challenges and novel approaches
- Hepatocellular carcinoma in the post-hepatitis C virus era: Should we change the paradigm?
- Honokiol-enhanced cytotoxic T lymphocyte activity against cholangiocarcinoma cells mediated by dendritic cells pulsed with damage-associated molecular patterns