First-principles study of the band gap tuning and doping control in CdSexTe1-x alloy for high efficiency solar cell∗
2019-08-16JingxiuYang杨竞秀andSuHuaiWei魏苏淮
Jingxiu Yang(杨竞秀) and Su-Huai Wei(魏苏淮)
1Department of Materials Science and Engineering,Jilin Jianzhu University,Changchun 130118,China
2Beijing Computational Science Research Center,Beijing 100193,China
Keywords: alloy,bowing effect,doping,II-VI semiconductors
1. Introduction
CdTe is one of the leading material for low-cost, highefficient, thin film solar cells due to its good optoelectronic property and the easy way to fabricate.[1]Although the power conversion efficiency (PCE) of the CdTe-based solar cell has so far reached to an impressive 22.1%, it is still much below the Shockley-Queisser limit (32%).[2]The current PCE in the world-record solar cell is mainly limited by the small open-circuit voltage (VOC), which is about 0.85 V compared to its band gap of 1.48 V at room temperature, as well as the relatively low short-circuit current (JSC), which reaches about 28 mA/cm2compared to JSC=30 mA/cm2under the Shockley-Queisser limit.[1,3]Currently, most efforts to improve CdTe-based solar cell efficiency have been trying to improve VOCinstead of the JSCbecause of the large deficiency in VOC. Some success has been achieved in increasing VOCby group V doping in CdTe.[4]However, it is still not clear whether such approach can obtain stable p-type absorbers because non-equilibrium doping process has to be used to improve the p-type doping.[5]On the other hand, one may increase the PCE by increasing JSC,which can be easily achieved by reducing the band gap of CdTe to harvest more long-wavelength sunlight. For example, if the band gap is reduced from 1.48 eV to 1.35 eV, the ideal JSCis increased from 30 mA/cm2to ∼36 mA/cm2. Because VOCof the current champion CdTe solar cell is still much lower than the band gap,[6,7]mainly due to the low carrier density in p-type CdTe,reducing the band gap of CdTe slightly, especially lowering the conduction band energy,is not expected to cause much decrease of the VOC.
Band gap tuning through alloying is widely used in semiconductors. Alloying CdTe at cation site could hardly achieve the reduction of the band gap, because the band gap always becomes wider when Cd is substituted by isovalent Zn,[8,9]and it is not desired to try alloying HgTe with CdTe given the toxicity of Hg. Therefore, one can only try to reduce the band gap of CdTe through alloying CdTe at anion site.The band gap of CdS and CdSe is 2.52 eV and 1.74 eV,respectively.[10]Although the band gap of CdS and CdSe are both larger than that of CdTe, alloying CdS or CdSe into CdTe can effectively reduce its band gap due to the large bowing effect.[11]Because the lattice mismatch between CdS and CdTe is large, the solubility of S into CdTe is low, which has been confirmed by previous theoretical and experimental studies.[11-13]Therefore, alloying CdTe with CdSe forming CdTe1-xSexseems to be the best choice to reduce the band gap effectively. Some of the recent experimental studies has already shown that diffusing CdSe into CdTe layer enables the increase of the JSC[2,14-16]However,so far,it is not clear how low the band gap of CdTe1-xSexcould be to maximize the increase of the JSC,although different experimental results regarding the CdTe1-xSexalloys have been reported.[17-21]
Furthermore, high p-type doping in CdTe is usually required for its solar cell performance, because as a minority carrier device, its electron mobility is much higher than the hole mobility. Although the dominant intrinsic p-type defect in CdTe isVCd,the obtained hole carrier density is too low for a good solar cell because VCdhas high formation energy. Therefore, extrinsic p-type dopants, such as CuCd, is often used in commercial CdTe-based solar cells.[22-24]However, it is also not clear how the formation of CdTe1-xSexalloy affects the doping properties in CdTe.
In this work, using the first principle hybrid-functional calculations,we find that the minimum of the band gap of the CdSexTe1-xalloy can approach 1.39 eV at about x=0.32,and most of the band gap reduction is through the lowering of the conduction band minimum. Our investigation of the doping property of the alloy reveals that the formation of the impurity CuCdexhibits dramatic bowing effect on the impurity formation energy,which can be utilized to improve the Voc,thus the PCE.The obtained band structure and the defect properties of the CdSexTe1-xalloy suggest that CdSexTe1-xalloy should be a better solar cell absorber than CdTe for the thin film solar cell application.
2. Computational methods
The first principle calculation in this work is performed by the VASP code.[25,26]PAW psuedopotentials with an energy cutoff of 350 eV were employed. PBEsol functional[27]with generalized gradient approximation (GGA) exchange correlation is used for the structure optimization of the bulk constitutes and alloys. All the atoms and the lattice vectors were fully relaxed until the force on each atom is less than 0.01 eV/˚A. For the defect calculation, the lattice vectors of the optimized alloy are fixed with all the atoms inside the supercell relaxed. To calculate the band structures and the band offsets,we have employed the hybrid functional[28]consists of 32%exact Hartree-Fock exchange mixed with 68%PBE exchange with spin-orbit coupling(SOC)to determine the band gap. This specific functional is chosen so that the calculated band gap of both zinc blende CdTe and CdSe are close to experimental values. Using the proposed functional, the calculated band gaps of zinc blende CdTe and CdSe are 1.52 eV and 1.69 eV,respectively, compared to the experiment values of 1.48 eV and 1.74 eV at room temperature.[10]The calculation of the band offsets of the series of CdSexTe1-xalloys follows the method described in our previous study.[11]
The CdSexTe1-xalloy is assumed to be random and is mimicked by the special quasirandom structures(SQS)[29]in the cubic supercell of 512 or 64 atoms,when x=0,0.25,0.5,0.75, and 1. The cubic supercell of 512 and 64 atoms are optimized with equivalent k-point sampling of 1×1×1 and 2×2×2,respectively. The averaged atomic correlation functions of the first neighbor pairs,triangles and tetrahedral of the SQS are the same as the perfect random alloys in the 512-atom supercells for all the mentioned concentrations. For the 64-atom supercell, the averaged atomic correlation functions of the first neighbored tetrahedral deviates from the perfect random alloys by 0.06 for x=0.25 and x=0.75 but is accurate enough for this case.The way to calculate the defect formation energy and the transition energy level is the same as stated in the previous work.[30-32]After testing with different functionals and supercells, the calculated formation energies are similar, and the calculated transition energy levels are converged to within 0.03 eV.Therefore, PBEsol functional and 64-atom supercells are adopted for the calculation of the doped alloys to reduce the computational cost.
3. Results and discussion
As described above, we have calculated the respective volume and mixing enthalpy ΔHmixof the random CdSexTe1-xalloys with Se composition x=0, 0.25, 0.5, 0.75, and 1 as shown in Figs. 1(a) and 1(b). The obtained lattice constant is 6.55 ˚A and 6.13 ˚A for pure CdTe and CdSe, respectively,in reasonably good agreement with experiment data.[10]As x increases, the volume of the CdSexTe1-xalloy decrease linearly due to the smaller size of Se, following the Vegard’s rule.[33]The ΔHmixof the random alloy is defined as the energy difference between the CdSexTe1-xalloy and the pure CdSe and CdTe with the corresponding ratio. The calculated ΔHmixcan be described quite well by the quadratic function Ωx(1-x),with the interaction parameter Ω =76.1 meV/f.u.Using the calculated value of Ω,the transition temperature is estimated to be 441 K, which is much lower than the experimental growth temperature,[16]therefore, it is easy to alloy CdSe into CdTe. In addition,the mixing enthalpy for x=0.25 is slightly lower than for x=0.75,reflecting the fact that it is easier to mix Se into CdTe than Te into CdSe.
The band gaps of the random CdSexTe1-xalloy are conventionally fitted to the equation:

where b is the bowing coefficient for the band gap. The hybrid functional calculated band gaps as function of the composition x are plotted in Fig.2(a),where the band gap bowing parameter b is found to be 0.725 eV and the band gap minimum is found at x=0.38.Given the slight difference of the calculated and experimental band gaps,the composition for the band gap minimum also slightly varies. Using the calculated bowing parameter b=0.725 and the experimental value of the band gaps at room temperature, the obtained band gap minimum of the random CdSexTe1-xalloy is predicted to be 1.39 eV at x=0.32,in agreement with a recent experiment result.[20]
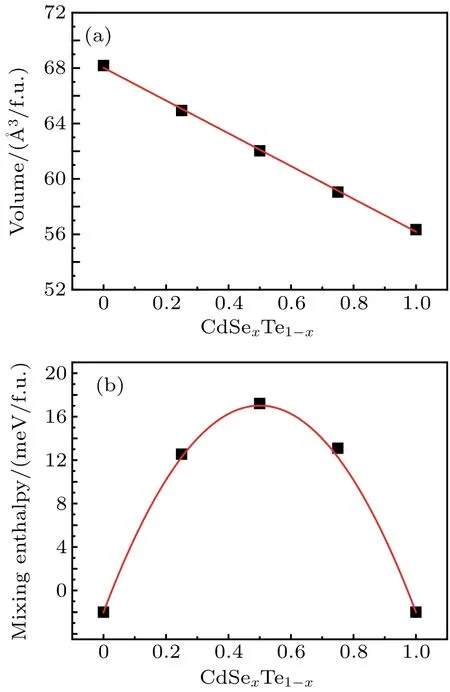
Fig. 1. The volume in units ˚A3/f.u. (a) and the mixing enthalpy ΔHmix in units meV/f.u. (b)as the function of the composition x for CdSexTe1-x alloys. The red lines in panels(a)and(b)are fitted curves.
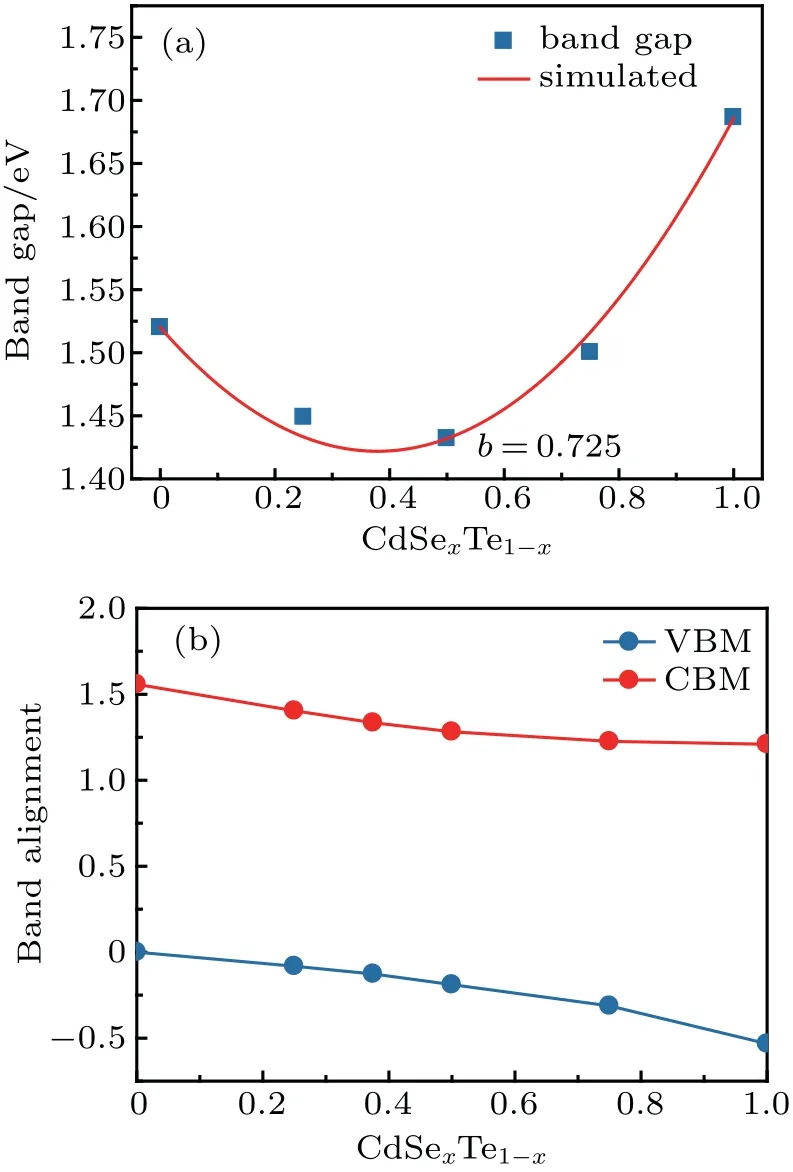
Fig.2.(a)The calculated band gaps as a function of x for CdSexTe1-x alloys,(b)The band alignments of the CdSexTe1-x alloys as a function of x.
The bowing of the band gaps for CdSexTe1-xalloys is caused by the bowing of both the band edges. As shown in Fig.2(b),the band offsets of the valence band minima(VBMs)and the conduction band minima(CBMs)between pure CdTe(x = 0) and CdSe (x = 1) are estimated to be 0.53 eV and 0.35 eV, respectively, consistent with the previous result.[11]Due to the strong intra valence band and intra conduction band coupling,the VBMs bow upwards and the CBMs bow downwards as x increases from 0 to 1,resulting the minimum band gap occurs at xmin. At this specific concentration, the CBM bows 0.09 eV more than the VBM does. The type-II band alignment between CdTe and CdSexTe1-xsuggests that a gradient CdSexTe1-xcell with Se-rich alloy in the front can help separate photogenerated electrons and holes, thus further improve the cell performance.
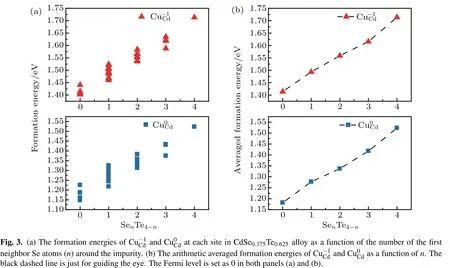
We first investigate the formation of the impurity CuCdin CdSe0.375Te0.625alloy modeled by a 64-atom SQS containing all five type Se4-nTen(n=0-4) nearest neighbor motifs around each Cd atom. The formation energy of CuCdunder Cd-rich condition at each possible site are calculated and plotted in Fig. 3(a). The formation energies of CuCdat charge state 0 and-1 depend mostly on the first neighbored configuration, although the farther neighbor configuration also has some effect,leading to the scattered formation energy within a given first neighbored motif. The averaged formation energies of the defect in different first neighbor motifs are shown in Fig.3(b). It is obvious that the averaged formation energy increases as the number of Se atoms increase in its first neighbor.As more Se atoms surround the impurity in the first neighbor motif, the bonding orbitals of the impurity contains more Se 4p orbitals,which has lower orbital energy[Fig.2(b)],thus,to form CuCdstate, it will cost more energy to create a hole.[34]The formation energy of Cu-1Cd(Fig.3(b)top)follows the trend of its neutral state(Fig.3(b)bottom),indicating the transition energy level ε(0/-1) for CuCdis less sensitive to its local configuration compared to the neutral formation energy. In other word, the CuCddefect is more like a delocalized defect in CdSexTe1-xalloys.
In alloys, the defect formation energy ΔHf(α, q, s, x) of defect α depends on charge state q, doping site s and the alloy composition x. To statistically investigate the defect property, it is more convenient to introduce an effective formation energy[35]ΔHeff(α,q,x,T),which is x-and T-dependent weighted average of the formation energy as given in Eq.(2),where kBis the Boltzmann constant, and N is the total number of the corresponding defect sites in alloys. To obtain the effective formation energy at charge states 0 and q, we could also define the effective transition energy level εeff(α,0/q,x,T)for defect α,which is the Fermi energy at which defect α at charge state 0 and q has the same effective formation energy as shown in Eq.(3).

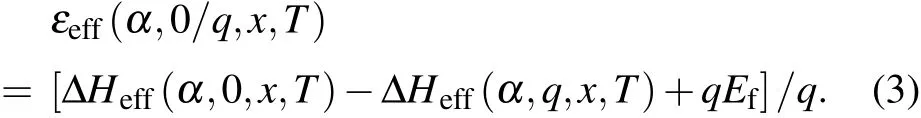
Considering the limit condition for the effective formation energy, equations(2)and(3)can be further deduced. At high temperature limit(T →∞),all the sites has equal weight,thus the effective formation energy ΔHeff(α,q,x,∞)is just the arithmetic average of the formation energies at all sites, so is the effective transition energy level εeff(α,0/q,x,∞).

On the other hand,at low temperature limit(T →0),only the site with the lowest formation energy at charge q(sq0)is occupied under equilibrium condition,so the effective formation energy ΔHeff(α,q,x,0)is just equal to ΔHf(α,q,sq0,x). The effective transition energy level εeff(α,0/q,x,0),therefore,is the energy difference between ΔHf(α,0,s00,x)and ΔHf(α,q,sq0,x). It is noted that the s00andmay not be at the same site.

The calculated effective formation energies for the defect CuCdat neutral and -1 states in CdSexTe1-xalloys (x=0,0.25,0.375,0.5,0.75,and 1)at the low temperature limit,the high temperature limit and a finite temperature T =600 K are shown in Figs. 4(a) and 4(b), respectively. It is interesting to see that the effective formation energy for CuCdimpurity exhibits a large bowing, i.e., they are much smaller than that of the composition averaged values in the pure CdTe and the pure CdSe. This is because, in addition to the electronic effect discussed above,the strain effect also plays an important role. The formation of CuCdcauses a compressive strain due to the smaller radius of Cu than Cd,thus the formation energy of CuCdwill be reduced as the local volume surround Cu is reduced.[36]This is the case when Cu is surrounded by Te and CuTe4cluster is compressed in the CdSexTe1-xalloy, so the formation energy of CuCdis much lower in the CdSexTe1-xalloy than in pure CdTe. The formation energy of CuCdalso decreases at the Se rich end when the CuTe4cluster is compressed most. At low temperature limit, Cu only occupy the lowest energy site(CuTe4cluster),so the bowing is the largest at the Se-rich side. At high temperature limit,the substitution occurs equally at all sites, so the effective formation energy change more smoothly as Se concentration increases. The formation of thegenerally follows the trend of Cu0Cdexcept that the bowing foris less dramatic than the bowing for CuCddue to the larger size of theimpurity.
As expected, the effective transition energy level increases as Se concentration increases in the alloy. It is interesting to see in Fig. 4(c) that at a given composition the effective transition energy level decreases as the temperature increase. This is because at the low temperature,the site with lower formation energy is preferentially occupied, where the impurity energy level for Cu0Cdis usually high to easily creating the hole. Therefore, the transition energy level(0/-1)is relatively high. At the high temperature limit,all the defect sites have nearly equal occupation probability,so the averaged effective transition energy is reduced. However, the variation of the effective transition energy is small at a given composition (∼0.04 eV), reflecting that CuCdis a relatively delocalized defect in CdSexTe1-xalloys.
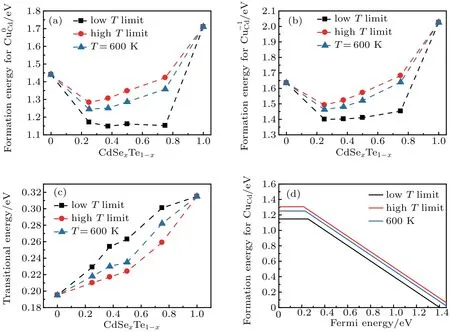
Fig. 4. The effective formation energies of Cu0Cd (a), Cu-1Cd (b) and the corresponding effective transition energy level (c) in CdSexTe1-x alloys(x=0, 0.25, 0.375, 0.5, 0.75, and 1) at the low temperature limit, the high temperature limit, and a finite temperature T =600 K. The Fermi level in panel(b)is set at 0. (d)The effective formation energy of CuCd as a function of the Fermi energy in the CdSe0.375Te0.625 alloy at the low temperature limit,the high temperature limit,and a finite temperature T =600 K.
The formation energy of the Cu0Cddefect in the CdSe0.375Te0.625alloy range from 1.31 eV to 1.15 eV at Cd-rich limit with the transition energy level varying from 0.217 eV to 0.254 eV, depending on the synthetic temperature,as shown in Fig.4(d). The insensitivity of the transition energy level and the lower formation energy of CuCdin the CdSe0.375Te0.625alloy suggests Cu doping in the alloy is more effective than that in pure CdTe.
4. Conclusion and perspectives
In summary, using first-principles calculations, we show that alloying CdTe with CdSe to form CdSexTe1-xalloys could be an effective approach to increase the PCE of the CdTe based thin film solar cells.The CdSexTe1-xalloy has two merits compared to CdTe: (i) reduced band gap (estimated to be 1.39 eV at x=0.32)to improve long-wavelength light harvest,thus improving JSC,(ii)lower formation energy of the shallow defect CuCdto improve the p-type conductivity, thus the potential to improve the Voc.
Acknowledgment
We acknowledge the computational support from the Beijing Computational Science Research Center.
杂志排行

Chinese Physics B的其它文章
- Lorentz transmission electron microscopy for magnetic skyrmions imaging∗
- Spin transport in antiferromagnetic insulators∗
- Non-Stokes drag coefficient in single-particle electrophoresis:New insights on a classical problem
- SymTopo: An automatic tool for calculating topological properties of nonmagnetic crystalline materials∗
- Tunable coupling between Xmon qubit and coplanar waveguide resonator∗
- Efficient solver for time-dependent Schrödinger equation with interaction between atoms and strong laser field∗