转移性前列腺癌生物信息学分析
2019-08-13黄东红陈桂芳
李 强,王 喻,黄东红,余 刚,陈桂芳
(1.中山火炬开发区医院泌尿外科,广东 中山 538437; 2.中山大学附属第三医院泌尿外科,广州 510630)
前列腺癌已成为全球癌症死亡的主要原因之一[1-3],随着人口老龄化及前列腺癌筛查的开展,前列腺癌的发病率将进一步升高。目前对于前列腺癌的筛查主要依靠前列腺特异性抗原(prostate-specific antigen,PSA),但其特异性不高且无法早期预测肿瘤转移[4]。另有研究表明PSA的筛查并不能明显降低前列腺癌死亡率[5-6],因此急需探索一种新型的前列腺癌早期诊断及转移预测标志物,以给予早期、精准的治疗。
肿瘤的发生、发展常伴随着成千上万个信号通路的改变,基础实验只能对其中有限的通路进行研究,无法整体、全面揭示肿瘤的生物学行为,而生物信息学分析结合了分子生物学与信息技术的优点,对测序数据进行全面、系统的分析,更好地诠释了肿瘤发生、发展的分子机制。特别是在二代测序技术的支持下,生物信息学分析现已飞速发展[7]。基于以上优点,本研究利用生物信息学分析对癌症基因组图谱数据库(The Cancer Genome Atlas,TCGA)和基因芯片公共数据库(Gene Expression Omnibus,GEO)进行分析,期望发现与前列腺癌转移相关的基因,为前列腺癌转移的早期诊断及治疗提供新的治疗靶点。
1 材料与方法
1.1测序数据及一般资料
1.1.1测序数据 前列腺癌转移相关基因数据主要从GEO(www.ncbi.nlm.nih.gov/)数据库中收录的相关芯片数据中挖掘。经过文献复习后最终选择GSE6919数据集作为前列腺癌转移基因数据集进行分析,该数据集包含GPL8300芯片平台采集的65个原发性肿瘤样本和25个转移样本。而前列腺癌差异分析数据则从TCGA(https://cancergenome.nih.gov/)中获取,包含52个正常样本和498个肿瘤样本。
1.1.2一般资料 选择2015年1月至2018年12月于中山火炬开发区医院及中山大学附属第三医院手术的前列腺增生及前列腺癌患者组织标本。前列腺增生症患者均接受了经尿道前列腺切除术,前列腺癌患者均接受了前列腺癌根治术或姑息性经尿道前列腺切除术,手术标本均经过病理学确诊。所有患者术前均未行内分泌治疗、化疗或放疗。
1.2方法
1.2.1数据预处理及差异表达 首先删除存在数据注释的样本及基因表达平均值低于1的基因数据,处理后的数据进行基因筛选。从TCGA和GEO数据库中获取前列腺癌测序数据,利用R 3.5.1软件及DESeq、heatmap2、ggplot2、clusterProfiler等R软件包进行分析。根据数据库信息将测序数据分为对照组(正常前列腺组织和前列腺增生组织),局限性前列腺癌组和转移性前列腺癌组,对数据进行过滤和标准化后进行差异性分析,其中差异基因的筛选条件为P<0.05,且log2FC≥2。
1.2.2基因模块的构建与筛选 将TCGA数据集中的差异基因和GSE6919中的差异基因取交集,得到的结果放回两个数据集中,分别使用corrplot软件包进行相关性分析,得出交集基因在两个数据集中的基因共表达模块,再将两个数据中最大模块的基因取交集,得出共表达系数最高的目的基因。
1.2.3差异表达基因的功能富集分析 利用R 3.5.1软件中Cluster Profiler软件包作基因本体论(Gene Ontology,GO)和京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)富集分析。本研究主要针对GO富集分析中的分子功能、生物过程和细胞组分3个模块以及KEGG通路进行分析。
1.2.4蛋白互作网络及关键基因筛选 使用String(https://string-db.org/)对目的基因进行蛋白互作网络分析,然后利用Cytoscape(http://www.cytoscape.org/)计算出关键基因并将其可视化。计算方法:使用MCC、MNC、Degree等进行计算,在所有算法中得分位于前3的基因视为关键基因。
1.2.5ACTA2的免疫组织化学检测 本研究收集前列腺增生患者30例,前列腺癌87例(局限性前列腺癌40例,转移性前列腺癌47例)的组织样本,使用免疫组织化学染色以明确ACTA2的表达情况。免疫组织化学操作参考说明书,首先使用ACTA2单抗(1∶1 000,Abcam lnc.,MA,ab220179)对切片进行4 ℃ 孵育过夜,接着二抗37 ℃孵育30 min后显色(Dako Diagnostics,Zug,Switzerland)。结果使用免疫组织化学染色评分(immunoreactive score,IRS)进行评估,IRS评分=染色程度×着色细胞百分比。染色程度:阴性0分,轻度1分,中度2分,重度3分;着色细胞百分比:0% 0分,<10% 1分,11%~50% 2分,51%~80% 3分,>80% 4分。IRS≥6分为高表达,<6分为低表达[8]。

2 结 果
2.1数据预处理及差异表达 TCGA包含52个正常样本,498个肿瘤样本,去除有注释样本,剩余44个正常样本,473个肿瘤样本;去除表达值低的基因后对剩余的31 314个基因进行分析。GSE6919用于分析的样本数为65个原发性肿瘤样本,25个转移样本,9 198个基因。对前列腺癌数据集进行差异性分析,获得前列腺癌中在转移和原发癌中差异表达的基因。其中GSE6919中转移和非转移差异表达基因共758个(上调370个,下调388个)。而TCGA数据集中差异表达基因共3 336个(上调1 161个,下调2 175个)。
2.2基因模块的构建与筛选 将两个数据集所获得的差异表达基因进行venny分析,结果显示前列腺癌转移的两个不同的数据集中,包含206个共同差异基因,GSE6919独有的基因552个,TCGA独有的基因3 130个,见图1。采用R 3.5.1软件中corrplot 软件包对上述206个基因进行相关性分析,结果显示在TCGA(图2A)和GSE6919(图2B)数据集中206个基因均主要构成3个模块,而TCGA最大模块与GSE6919最大模块存在14个共有基因,见表1。
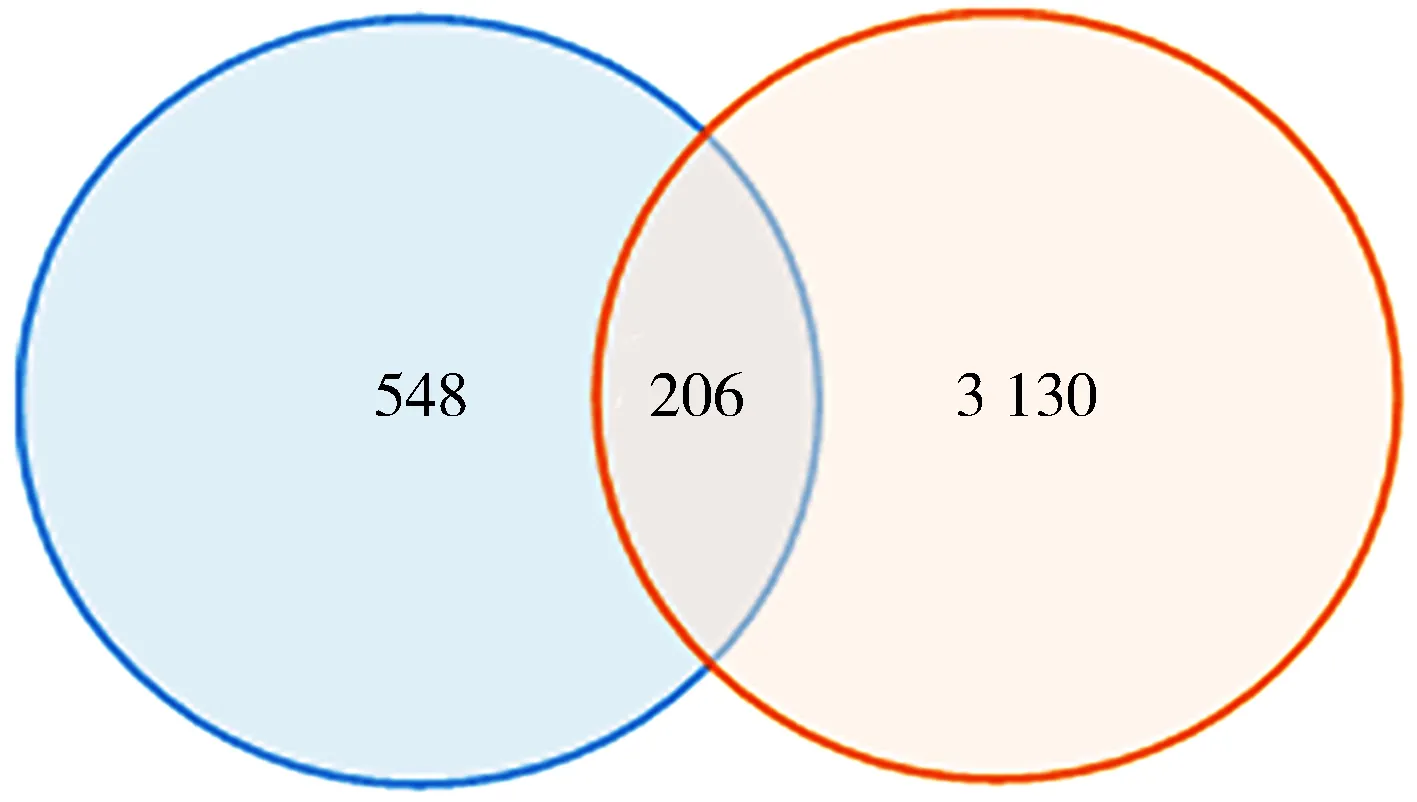
图1 TCGA与GSE6919交集基因韦恩图
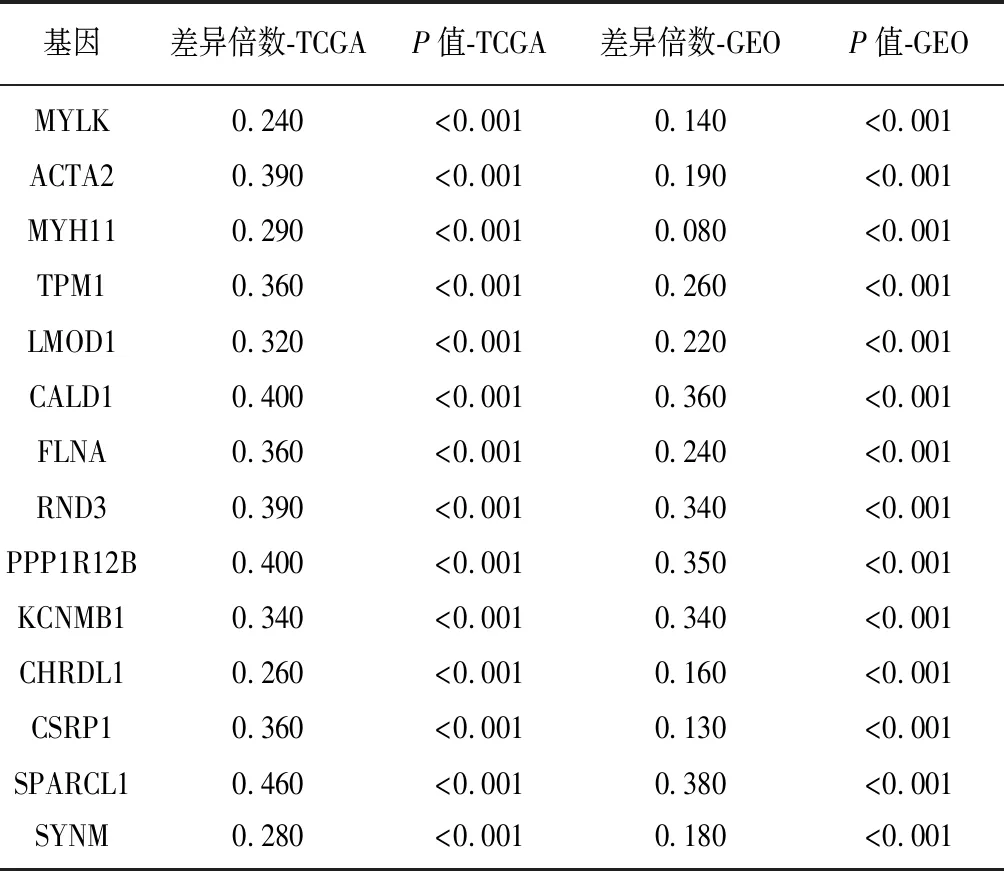
基因差异倍数-TCGAP值-TCGA差异倍数-GEOP值-GEOMYLK0.240<0.0010.140<0.001ACTA20.390<0.0010.190<0.001MYH110.290<0.0010.080<0.001TPM10.360<0.0010.260<0.001LMOD10.320<0.0010.220<0.001CALD10.400<0.0010.360<0.001FLNA0.360<0.0010.240<0.001RND30.390<0.0010.340<0.001PPP1R12B0.400<0.0010.350<0.001KCNMB10.340<0.0010.340<0.001CHRDL10.260<0.0010.160<0.001CSRP10.360<0.0010.130<0.001SPARCL10.460<0.0010.380<0.001SYNM0.280<0.0010.180<0.001
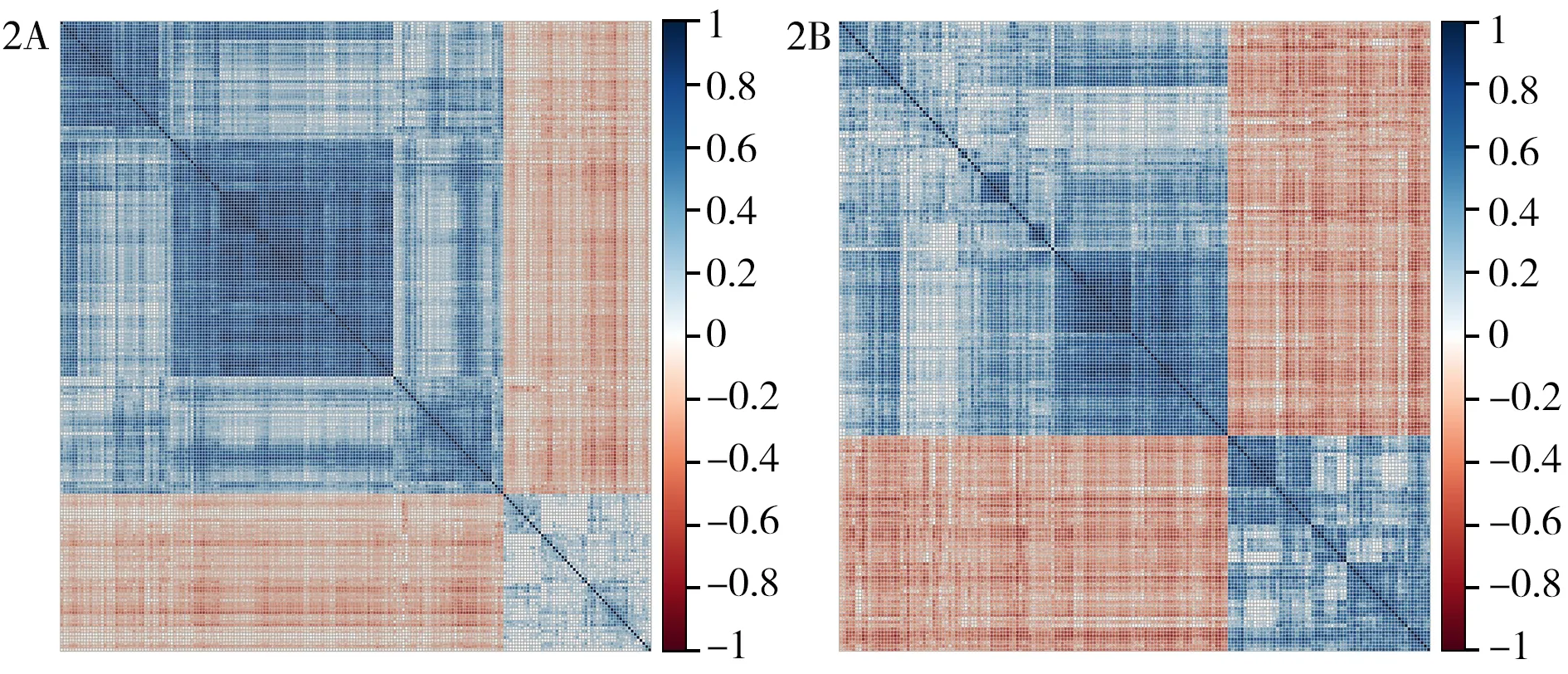
图2 交集的差异基因在TCGA数据集(2A)和
2.3差异表达基因的富集分析 对TCGA和GSE6919数据集中的最大模块基因进行富集分析,见图3,然后对14个交集基因进行单独的富集分析,结果提示这14个基因主要富集到血管平滑肌收缩、局部黏着、Apelin信号通路、催产素信号通路、环鸟苷酸蛋白激酶G信号通路中,见图4。

3A和3D:分子功能; 3B和3E:生物过程; 3C和3F:细胞组分; 3G和3H:KEGG通路分析

4A:分子功能;4B:生物过程;4C:细胞组分;4D:KEGG通路分析
2.4蛋白互作网络及关键基因筛选 对14个目的基因进行蛋白互作网络分析,结果显示其中10个基因可能存在相互联系:MYLK、ACTA2、MYH11、TPM1、LMOD1、CALD1、FLNA、RND3、PPP1R12B、KCNMB1。然后使用Cytoscape软件对其进行可视化和关键基因的筛选,结果提示MYH11、MYLK和ACTA2是这10个基因的关键基因,见表2,调控着大多数的蛋白见图5。

表2 关键基因的计算结果

图5 蛋白互作网络分析
2.5免疫组织化学 在前列腺癌组织中,ACTA2的表达较前列腺增生明显降低,ACTA2的表达随前列腺癌的进展逐渐下降,见图6。前列腺增生组、局限性前列腺癌组、转移性前列腺癌组的IRS分别为(7.43±0.49)分、(4.85±0.22)分、(2.66±0.27)分,各组比较差异有统计学意义(F=223.790,P<0.001),局限性前列腺癌组、转移性前列腺癌组的IRS评分低于前列腺增生组,转移性前列腺癌组低于局限性前列腺癌组(P<0.01)。Gleason评分≥7分的患者ACTA2低表达率高于Gleason评分<7分的患者(P<0.05);转移性前列腺癌患者ACTA2低表达率高于局限性前列腺癌患者(P<0.05);不同年龄、PSA水平和病理分期患者的ACTA2低表达率比较差异无统计学意义(P>0.05)。见表3。
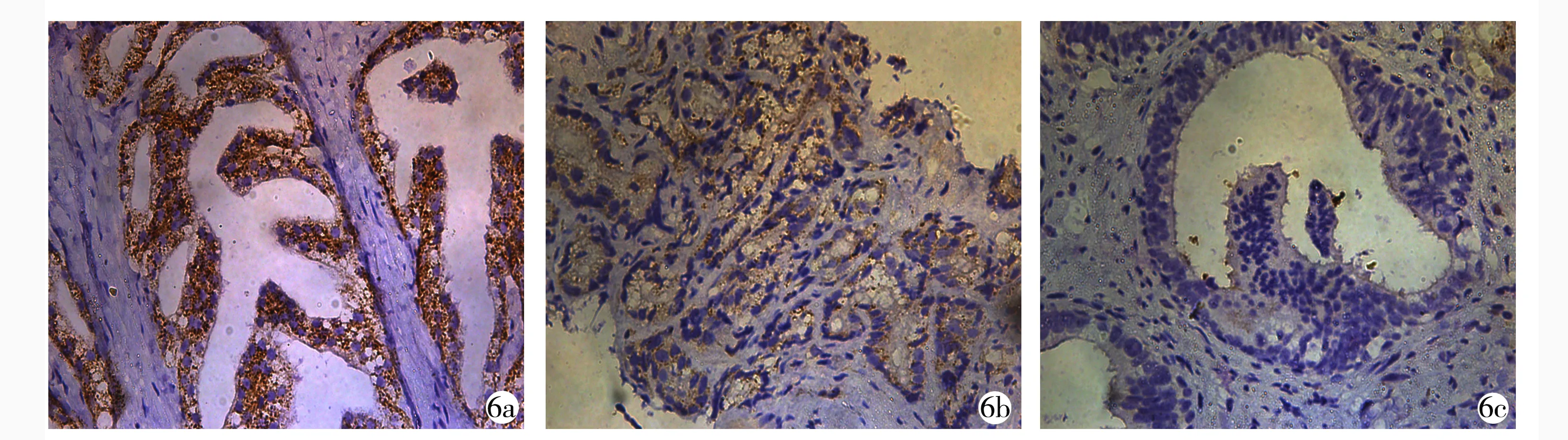
6a:前列腺增生; 6b:局限性前列腺癌; 6c:转移性前列腺癌

表3 前列腺癌患者各临床指标ACTA2表达水平比较 [例(%)]
PSA:前列腺特异性抗原
3 讨 论
美国国家癌症研究所公布的研究结果显示,任何肿瘤的发生、发展都伴随着一系列基因的高频突变,并且相互间存在复杂的基因调控网络[9]。传统的分子生物学实验只能研究有限的基因功能及通路,而作为集合了分子生物和信息技术优点的生物信息学分析可研究多基因调控事件在肿瘤发生、发展中的作用。
本研究从公共数据库获取前列腺癌相关数据,经过一系列的分析最终发现14个与前列腺癌转移相关的基因,上述基因主要富集到血管平滑肌收缩、局部黏着、Apelin信号通路、催产素信号通路、环鸟苷酸蛋白激酶G信号通路中,提示上述基因可能通过这些通路促进前列腺癌的发生、发展。使用String对这14个基因进行蛋白互作网络分析,结果提示其中10个基因存在相互作用,并且MYLK、ACTA2、MYH11可能发挥关键调控作用。MYH11广泛存在于哺乳动物中,对机体的信号转导、肌肉收缩及细胞器运动起调控作用。Wang等[10]研究发现,MYH11在结肠癌中低表达,且表达水平与肿瘤分期相关。Lu等[11]研究证明MYH11在膀胱癌中表达降低,且表达水平与患者肿瘤侵袭程度呈负相关。MYLK是一种钙调蛋白依赖性苏氨酸-丝氨酸激酶,广泛存在于各种真核细胞及非肌肉性细胞,属于免疫球蛋白超家族[12]。MYLK主要通过增强肌球蛋白酶活性促进肌球蛋白收缩和应力纤维的形成、黏着,最终导致细胞的迁移和分裂[13-14]。Minamiya等[15]首次报道了在复发转移的非小细胞肺癌患者中MYLK mRNA的表达水平高于无复发转移患者。Chen等[16]研究结果显示MYLK mRNA的表达上调可调控胃癌细胞增殖、凋亡、侵袭与转移。而Lee等[17]报道在结肠癌中MYLK mRNA相对于正常组织下调,本研究结果与Lee等[17]的研究结果一致。目前MYLK的下调机制仍不清楚,可能与DNA突变、MYLK假基因有关[18]。ACTA2主要分布于肌细胞,是收缩器的主要成分[19]。Jeon等[20]报道表皮生长因子和人类表皮生长因子受体2二聚体可调控ACTA2的表达进而影响乳腺癌的侵袭和转移。Lee等[21]报道ACTA2可调控c-MET和FAK进而促进肺腺癌的转移。可见,MYLK、MYH11和ACTA2均与平滑肌细胞功能相关,可能通过影响细胞的黏附、侵袭能力,进而导致前列腺癌的转移。
为进一步验证上述结果,本研究纳入前列腺增生及前列腺癌患者共117例进行ACTA2免疫组织化学分析,免疫组织化学与上述研究结果一致。随着前列腺癌的进展,ACTA2的表达也逐渐下降,当患者出现转移时ACTA2的表达明显降低。本研究结果显示,Gleason评分≥7分的患者ACTA2低表达率高于Gleason评分<7分的患者(P<0.05),转移性前列腺癌患者ACTA2低表达率高于局限性前列腺癌患者(P<0.05),表明ACTA2的下调可导致前列腺癌的进展。
综上所述,MYLK、MYH11和ACTA2可能是导致前列腺癌转移的关键基因,上述基因参与平滑肌细胞功能的调节,具有调控细胞的黏附、侵袭等功能。另外,本研究还证实ACTA2的下调与前列腺癌进展相关,提示该基因可能成为新的前列腺癌诊断标志物和治疗靶点。
