Drug Side-Effect Prediction Using Heterogeneous Features and Bipartite Local Models
2019-08-13YihengWentaohaoChengchengSunandQianLi
Yi ΖhengWentao ΖhaoChengcheng Sun and Qian Li
Abstract: Drug side-effects impose massive costs on society,leading to almost one-third drug failure in the drug discovery process.Therefore,early identification of potential side-effects becomes vital to avoid risks and reduce costs.Existing computational methods employ few drug features and predict drug side-effects from either drug side or side-effect side separately.In this work,we explore to predict drug side-effects by combining heterogeneous drug features and employing the bipartite local models (BLMs)which fuse predictions from both the drug side and side-effect side.Specifically,we integrate drug chemical structures,drug interacted proteins and drug associated genes into a unified framework to measure the comprehensive similarity between drugs first.Then,high-quality and balanced training samples are selected for individual drugs and individual side-effects using the designed balanced sample selection framework,based on drug comprehensive similarities and side-effect cosine similarities respectively.Trained with corresponding training samples,BLMs first predict drugs associated with a given side-effect,then predict side-effects for a given drug.This produces two independent predictions for each putative drug-side-effect association which are further combined to give a definitive prediction.The performance of the proposed method was evaluated on side-effect prediction for 901 drugs from DrugBank.Particularly,we performed 5-fold cross-validation experiments on the 742 characterized drugs and independent testing experiment on the 159 uncharacterized drugs.The simulative predictions show that the side-effect prediction performance is significantly improved owing to the integration of information from drug chemical,biological and genomic spaces,the proposed sample selection framework,and the implemented BLMs.
Keywords: Side-effect prediction,heterogeneous features,bipartite local models.
1 Introduction
Drug side-effects impose massive costs on society,resulting in significant morbidity and mortality.They are estimated to be the fourth leading cause of death in the United States,responsible for around 100,000 deaths each year [Giacomini,Krauss,Roden et al.(2007);Zheng,Peng,Zhang et al.(2018);Zheng,Peng,Ghosh et al.(2019)].Side-effects also account for around one-third of drug failures in the drug discovery process [Kennedy(1997)].The early identification of potential side-effects,before reaching the clinical stages,is of critical importance.
Wet experiments (e.g.,preclinicalin vitrosafety profiling) are capable to identify potential side-effects,but such experimental identification remains challenging in terms of the cost and efficiency.Recently,severalin silicoprediction methods have been proposed to improve this expensive and time-consuming process.These methods employed different drug features and prediction models,aiming to achieve efficient and accurate predictions.
Drug chemical structures are widely used as features to make side-effect predictions.In 2007,Bender et al.first attempted to predict side-effects across hundreds of categories from drug chemical structures alone,and demonstrated the feasibility of using drug chemical structures for side-effect prediction [Bender,Scheiber,Glick et al.(2007)].Pauwels et al.performed sparse canonical correlation analysis on correlated sets of drug chemical substructures and side-effects to predict side-effects [Pauwels,Stoven and Yamanishi (2011)].They showed the usefulness of their method via predicting 1385 sideeffects in SIDER.Target proteins and pathways are also employed as features to predict potential side-effects.Iwata et al.systematically analyzed the correlation between sideeffects and protein domains based on drug-target interaction network [Iwata,Mizutani,Tabei et al.(2013)].They showed that the inferred side-effect-domain association network was useful for estimating common drug side effects.Drug target data and clinical observation data were combined to develop a computational framework for accurate side-effect prediction of trial drugs [Huang,Wu and Chen (2011)].The authors figured out that gene annotation information of target proteins could increase the prediction accuracy.Fukuzaki et al.leveraged cooperative pathways and gene expression profiles to predict side-effects [Fukuzaki,Seki,Kashima et al.(2009)].However,their method depends heavily on the availability of gene expression data.Instead of using a single drug feature (e.g.,drug chemical structure),researchers tried to predict drug sideeffects by integration of different types of drug features [Yamanishi,Pauwels and Kotera(2012);Zhang,Chen,Tu et al.(2016)].Yamanishi et al.[Yamanishi,Pauwels and Kotera(2012)] integrated drug chemical structures and drug target proteins in a unified framework for side-effect prediction.Extensive experiments demonstrated that the prediction performance was significantly improved owing to the integration.Analogously,impacts of different combination of drug features were investigated in Zhang et al.[Zhang,Chen,Tu et al.(2016)].Compared with methods based on a single drug feature,all feature integration methods produced better performances.
Recently,supervised learning with bipartite local models has been demonstrated to give superior performance to precursor algorithms in predicting drug potential targets [Mei,Kwoh,Yang et al.(2012);Bleakley and Yamanishi (2009);Xiang,Li,Hao et al.(2018)].It combines predicted targets of a given drug and predicted drugs which target a given target to achieve precise predictions.
Taking advantages of feature integration and BLMs,we propose to predict potential drug side-effects based on integration of drug chemical structures,drug interacted proteins and drug associated genes using BLMs.First,we integrate the chemical space of drug chemical structures,biological space of drug interacted proteins and genomic space of drug associated genes into a unified framework to measure similarities between drugs.Then,similarities between side-effects are calculated by the cosine similarity measurement.Next,a high-quality and balanced training sample set is selected for each drug and each sideeffect according to corresponding drug comprehensive similarities and side-effect cosine similarities respectively.Finally,BLMs predicts each potential drug-side-effect association by combining prediction results from the drug side and the side-effect side.We demonstrated the usefulness of the proposed method on simulative prediction of associations between 901 drugs and 635 side-effects.Specifically,742 characterized drugs whose sideeffect profiles are available in SIDER were used for 5-fold cross validation experiments.The remaining 159 uncharacterized drugs whose side-effects are not stored in SIDER,were used for independent testing experiment.Both the cross-validation experiments and the independent testing experiment show that the prediction performance improves steadily owing to the integrated drug features,selected high-quality samples and the BLMs.
2 Methods
2.1 Data resources
2.1.1 Data set of drug source
In this work,we focus on 901 drugs which are classified as “small molecules” in Law et al.[Law,Knox,Djoumbou et al.(2014)],a comprehensive drug database.Drug information including drug names,drug SMILES strings,drug interacted proteins,and ChEMBL ids was extracted from DrugBank.The protein-Gene Ontology (GO) association information was downloaded from the EMBL-EBI website [Harris,Clark,Ireland et al.(2004)].The drug-disease (or drug-indication) associations were obtained via searching the ChEMBL online database using corresponding ChEMBL ids [Bender (2010)].And the disease-gene association data were downloaded from CTD [Davis,Grondin,Johnson et al.(2017)].
2.1.2 Data set of drug-side-effect associations
The drug-side-effect association information was obtained from SIDER [Kuhn,Letunic,Jensen et al.(2015)].Some rare side-effects are associated with very few drugs.Little information can be provided from them.Therefore,we removed side-effects which were associated with less than 30 drugs.Finally,we obtained a data set of 901 drugs,635 sideeffects,and 73,295 associations.The side-effect list and drug list can be found in Tab.S1 and S2 in Additional file 2.The 901 drugs are consisted of 742 characterized drugs whose side-effect profiles are available in SIDER,and 159 uncharacterized drugs whose associated side-effects are not available in SIDER.
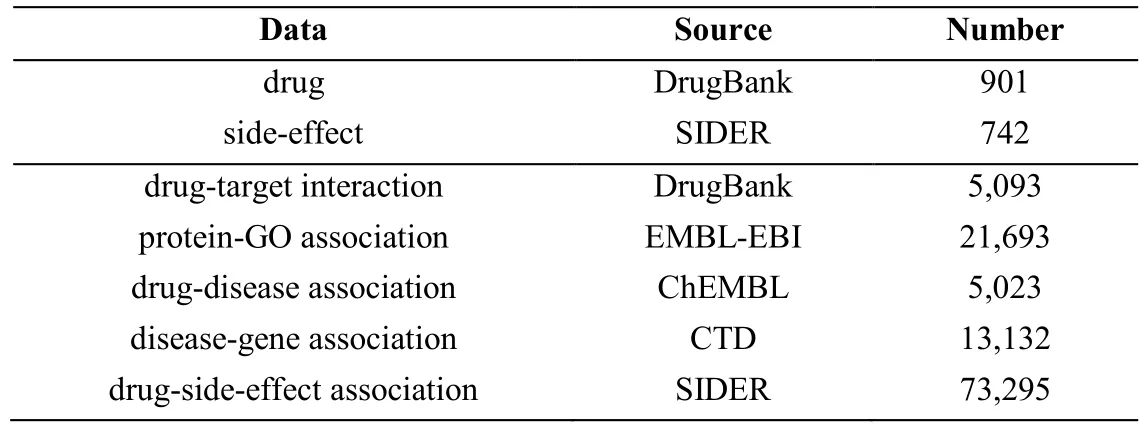
Table1:Summary of the data resources
2.2 Prediction with BSSF and BLMs
The framework of the proposed method is illustrated in Fig.1.It includes steps for the calculation of drug similarity and side-effect similarity,sample selection,data representation,and prediction models.

Figure1:The framework for drug side-effect prediction.The framework consists of four parts,namely similarity calculation,sample selection,data representation and bipartite local models.DrugBank:drug data;EMBL-EBI:gene ontology data;ChEMBL:drugdisease association data;CTD:disease-gene association data;SIDER:side-effect resource;ASCs:accumulative similarity scores
2.2.1 Drug similarity calculation
A.Similarity of chemical structures
The drug chemical structure was converted into a fingerprint by the Chemistry Development Kit (CDK) [Steinbeck,Hoppe,Kuhn et al.(2006)] from its SMILES string.
The fingerprint is an 881-dimensional binary vector,where each element encodes the presence or absence of the PubChem substructure by 1 or 0 [Chen,Wild and Guha(2009)].Then,the chemical structure similarity between two drugs was calculated as the Tanimoto 2D score between their fingerprints.For drug djand drug dk,their chemical structure similarity score is given by:

where ∧ and ∨ are bitwise “and” and “or” operators respectively;fljand flkare the ltℎbit of fingerprints of drug djand drug dkrespectively.
B.Similarity of interacted proteins
The similarity between two proteins was calculated as the overlapping rate of their associated GO terms.Let GOmand GOnbe the GO term set for protein pmand pnrespectively,the similarity score between them is defined as:

where ∩ and ∪ are “intersection” and “union” operators respectively.Zhang et al.demonstrated the integration of drug interacted proteins including drug targets,drug enzymes,and drug transporters could improve the prediction performance [Zhang,Chen,Tu et al.(2016)].Therefore,we combined them with drug carriers together to compute the drug interacted protein similarity.The interacted protein similarity score between drug djand drug dkis computed by:

where Njand Nkare the total number of proteins in the interacted protein sets of drug djand drug dkrespectively.
C.Similarity of associated genes
The drug associated gene similarity is measured by genes associated with drug indications (i.e.,diseases).We leverage the method proposed in Cheng et al.[Cheng,Li,Ju et al.(2014)] to measure the similarity between two diseases.First,the information content (IC) of a disease dis from Disease Ontology (DO) [Schriml,Arze,Nadendla et al.(2011)] is calculated as follows:

where p(dis) equals the number of genes associated with disease dis divided by the total number of genes related to DO.Then the similarity between two diseases is defined as the IC of their most informative common ancestor (MICA).MICA is the common ancestor which has the maximum IC.We constructed DO based on the tree view of diseases in the MeSH Browser [Lipscomb (2000)].The DO contains 4,578 disease terms and 11,480 “is_a” relationships among terms.To keep consistent with chemical structure similarity and interacted protein similarity,the similarity between two diseases is normalized to the range [0,1] as follows:

where Sdis(dism,disn) and NSdis(dism,disn)are the original disease similarity and normalized disease similarity between disease dismand disease disnrespectively;andarethe maximum and minimum similarity among all disease pairs respectively.Then theassociated gene similarity is definedas:

where Njand Nkare the total number of indications of drug djand drug dkrespectively.D.Integration of drug similarity“Mean” is used as the consensus similarity inference method to integrate the above three measurements of drug similarities into a single comprehensive similarity.
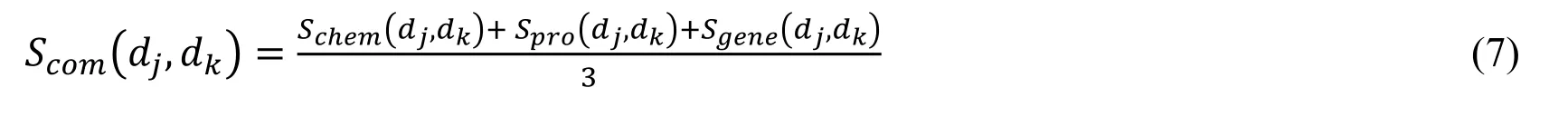
2.2.2 Side-effect similarity calculation
Side-effects are represented as 742-dimensional vectors using their associations with the 742 characterized drugs.Each element of the vector encodes the presence or absence of the corresponding side-effect-drug association by 1 or 0.The similarity between two side-effects is defined as the cosine angle between their vectors.
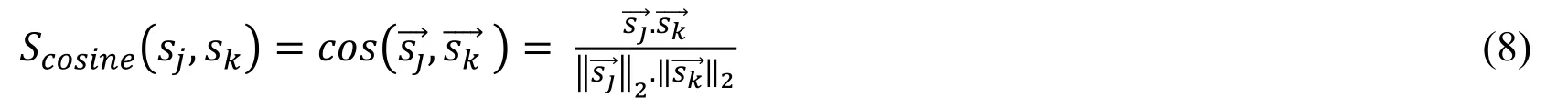
2.2.3 Sample selection framework
In the side-effect prediction task,the number of positive samples and the number of negative samples are usually imbalanced.Besides,the prediction performance is severely impeded by the lack of reliable negative samples and high-quality positive samples.In this work,we developed a framework to select balanced high-quality training samples.We took the selection of training drug samples for a side-effect sjas an example to illustrate the selection process:
A.Obtaining the smaller number ns,between the labeled drug number (Nl) and the unlabeled drug number (Nu).Labeled and unlabeled drugs are drugs which are known to associate with sjin SIDER or not respectively.
B.If Nl ≥ Nu,then compute the accumulative similarity score (ASC) between each labeled drug and all unlabeled drugs.For the labeled drug di,its ASC is calculated as follows:

Otherwise compute the ASC between each unlabeled drug and all labeled drugs analogously.
C.If Nl ≥ Nu,then sort labeled drugs in ascending order according to their ASCs.The top Nu labeled drugs and all the unlabeled drugs are selected as training drug samples for sj.Otherwise,sort unlabeled drugs in ascending order.The top Nl unlabeled drugs and all labeled drugs are selected as training drug samples for sj.
2.2.4 Data representation
Each drug is represented as a 742-dimensional vector.The elements encode for the comprehensive similarity between the drug and the 742 characterized drugs.In a similar way,each side-effect is represented as a 635-dimensional vector whose elements denote the cosine similarity between the side-effect and all side-effects.
2.2.5 Bipartite graph inference with local models
The drug-side-effect associations can be viewed as a bipartite network,in which vertexes are drugs and side-effects,edges are their associations.Thus,the problem of predicting new drug-side-effect associations is to infer new edges from the bipartite network.We adopt the idea proposed in Bleakley et al.[Bleakley and Yamanishi (2009)] to train several local models to predict new edges from both the drug side and side-effect side.Specifically,the presence or absence of edge eijbetween drug diand side-effect sjis predicted as follows:
A.Drug side
(1) Excluding drug di,we obtain the selected training drug samples for side-effect sj.Among the training drug samples,drugs known to have sjare labeled as +1 and the rest are labeled as -1.
(2) Vectors of all training drug samples and their labels are fed into an auto-classifier for training.
(3) The trained classifier is employed to predict the label of drug di.
B.Side-effect side
(1) Excluding side-effect sj,we obtain the selected training side-effect samples for drug di.Among the training side-effect samples,side-effects which are known to be associated with drug diare labeled as +1,otherwise -1.
(2) An auto-classifier is trained using vectors of all training side-effect samples and their labels.
(3) The trained classifier is leveraged to predict the label of side-effect sj.
C.Prediction result integration
Prediction from the drug side and side-effect side using different datasets provides two independent predictions of the same edge (i.e.,association).The two predictions are combined to give a definitive prediction using their average value average{x,y},where x and y are the predicted scores from drug side and side-effect side respectively.
3 Results
3.1 Performance evaluation and comparison
We employed the k-nearest neighbor (KNN) as the local auto-classifier for prediction.K was set as one-third of the number of training samples (rounded down).We tested nine approaches:(1) BSSF_BLM (2) Random_BLM (3) Original_BLM,(4) BSSF_SS,(5)Random_SS,(6) Original_SS,(7) BBSF_DS,(8) Random_DS,and (9) Original_DS on their abilities to predict known drug-side-effect associations using the 742 characterized drugs.BSSF indicates the approaches are based on the proposed balanced sample selection framework;Random denotes the samples are selected randomly;Original means the approaches are performed without any sample selections;BLM implies the approaches employ the bipartite local models;DS and SS suggest the prediction are made using information from the drug side and the side-effect side respectively.The comprehensive drug similarity and cosine side-effect similarity were applied to measure the similarity between drugs and similarity between side-effects respectively in all the nine approaches.The performance is evaluated by the 5-fold cross validation:(1)Samples in the gold standard are split into 5 roughly equal-sized subsets;(2) Each subset is taken in turn as the test set,the remaining four subsets are used as training set;(3) All results over the 5-fold validation are used for evaluation.The receiver operating characteristic (ROC) curve,precision-recall (PR) curve,the area under ROC curve (AUC)and the area under the precision-recall curve (AUPR) are used as the evaluation metrics.To obtain robust results,approaches based on randomly selected samples were repeated 5 times and the average results were used for evaluation.
Fig.2 shows the global ROC curves for the above nine approaches based on 5-fold cross validation experiments,where all the predicted scores for each drug-side-effect association were merged and a global curve was drawn for each approach.The resulting AUC scores for the nine approaches are 0.9879,0.9688,0.8998,0.9763,0.8500,0.9075,0.8973,0.8175,and 0.6434 respectively.It seems that approaches based on the proposed BSSF performed better than those based on randomly selected training samples and raw training samples.For example,BSSF_DS outperformed Random_DS and Original_DS at 0.0798 and 0.2538 respectively.This result demonstrates the feasibility of the proposed sample selection framework.Approaches employ the bipartite local models seem to work better than approaches solely based on prediction from the drug side or side-effect.For instance,compared with BSSF_DS and BBSF_SS,BSSF_BLM achieved 0.0906 and 0.0116 higher AUC score respectively.It suggests the integration of predictions from both the drug side and side-effect side is meaningful.Among the nine approaches,BSSF_BLM which employs both the proposed balanced sample selection framework and bipartite local models achieved the best performance.The same results can be observed from the global PR curves (see Fig.S1 in Additional file 1).
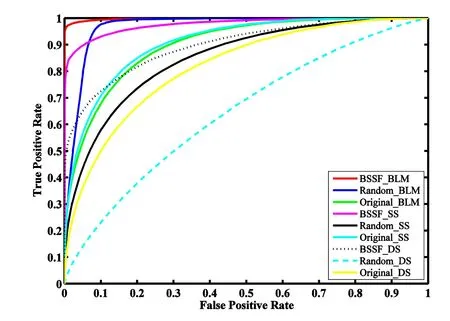
Figure2:ROC curves based on the 5-fold cross validation.Comparison of the performance among 9 approaches.BSSF indicates the approaches are based on the proposed balanced sample selection framework;Random denotes the samples are selected randomly;Original means the approaches are performed without any sample selections;BLM implies the approaches employ the bipartite local models;DS and SS suggest the prediction are made using information from the drug side and the side-effect side respectively
To demonstrate the significance of integrating gene information (i.e.,drug associated gene similarity),we investigated the prediction accuracy for each drug and each sideeffect with a high level of confidence.We compared these results with one state-of-theart work which integrates chemical structures and target proteins into a unified framework (hereinafter refer to as ChemBio) [Yamanishi,Pauwels and Kotera (2012)].Fig.3 illustrates the boxplots which represent the distribution of area under the PR curve(AUPR) for individual drugs and side-effects respectively.Compared to ChemBio,the proposed approaches integrated more drug interacted proteins (i.e.,enzymes,transporters and carrier) and drug associated genes.Predicting side-effects for small molecule drugs in DrugBank,the proposed approaches significantly outperformed ChemBio,which demonstrates the high-performance prediction power of the proposed methods.In addition,it indicates that the integration of gene information and more drug interacted proteins makes sense.
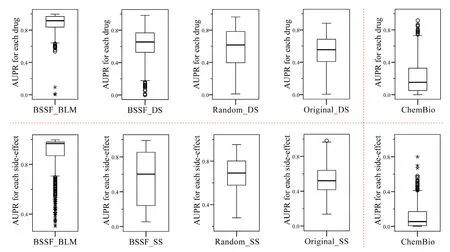
Figure3:Boxplots of the AUPR (area under the precision-recall curve) scores for each drug and each side-effect.Comparison of performances among the proposed approaches and the state-of-the-art work “ChemBio” [Yamanishi,Pauwels and Kotera (2012)].The upper five boxplots show the AUPR scores for individual drugs,and the five boxplots below illustrate the AUPR scores for individual side-effects
To directly show how well the proposed approaches work,we also investigated the number and ratio of drugs whose top ranked predicted side-effects are confirmed in SIDER.Related results are listed in Tab.2.Consistent with previous results,BSSF_BLM achieved the best performance.For example,the proposed approach BSSF_BLM ranked known side-effects in all top 10 predicted results for 653 drugs (82.08%) of the 742 characterized drugs and ranked known side-effects in all top 50 predicted results for 302 drugs (40.7%).These results further demonstrate the prediction power of the proposed method.
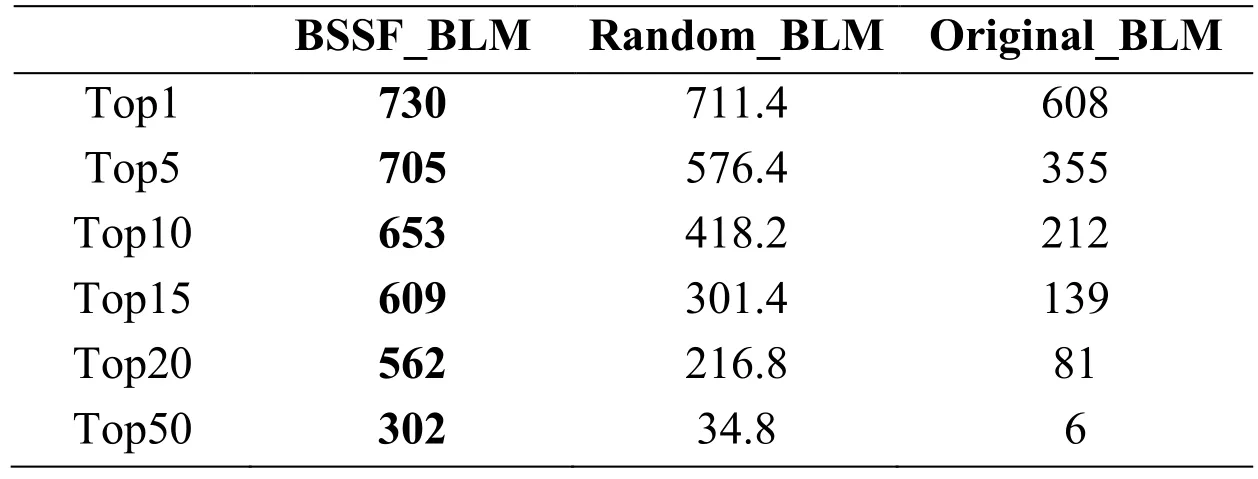
Table2:Performance statistics of the top predicted drug-side-effect associations
Top x indicates the number of drugs,whose top x ranked predicted side-effects all are known in SIDER.The best result in each row is highlighted in bold.Approaches based on randomly selected samples were repeated 5 times and the average results were presented.
3.2 Side-effect prediction for uncharacterized drugs
In DrugBank,there are still 159 uncharacterized drugs whose chemical structure information,interacted protein information and associated gene information are available,but their side-effect profiles are not stored in SIDER.We conducted the independent testing experiment using BLM_DS on them.All the 742 characterized drugs were used as training set.The whole prediction results are reported in Tab.S3 in Additional file 2.It is not practical to analyze all predictions,so we focused on predicted side-effects of uncharacterized drugs related to brain-diseases or withdrawn from the market.We tried to confirm some predicted drug-side-effect associations using other sources,such as DrugsCom and PubMed.
Acetylsalicylic acid (DB00945) is the only uncharacterized drug prescribed for brain diseases.It is used to treat Transient Cerebral Ischemia (TCI) which is a subclass of Brain Ischemia [Law,Knox,Djoumbou et al.(2018)].TCI attacks around 30% to 40% patients who have stroke,a major cause of disability and death in North America [Cusimano and Ameli (1989)].Consequently,the treatment of TCI is beneficial in preventing stroke.The side-effect information of “Acetylsalicylic acid” is not available in SIDER,however,the information can be found in DrugsCom [Drugs.com (2018)].So it is reasonable to evaluate the prediction performance by analyzing how its side-effects in DrugsCom were predicted.The side-effect names are not always consistent between side-effects from DrugBank and DrugsCom when referring the same side-effects.So we mapped sideeffects of DrugsCom into side-effects of DrugBank.The mapping results as well as the prediction scores can be found in Tab.S4 in Additional file 2.58 side-effects are reported in DrugsCom.Among them,45 side-effects are successfully mapped into side-effects of DrugBank and 24 side-effects are of high credibility.Fig.4 illustrates the 24 highcredibility side-effects and their prediction scores.It can be seen that 16 out of 24 highcredibility side-effects obtained prediction scores larger than 0.5 (common threshold).34 out of the 45 mapped side-effects (75.56%) were successfully predicted.
There are 10 withdrawn drugs whose side-effect profiles are not available in SIDER.We investigated how much our approach could explain why they were withdrawn from the market based on literature evidences.Part of the serious side-effects and their corresponding predicted scores are listed in Tab.3.The entire results and detailed evidences are listed in Tab.S5 in Additional file 2.From Tab.S5,it can be seen that most side-effects were successfully captured.For instance,cisapride (DB00604),a gastroprokinetic agent increases motility in the upper gastrointestinal tract was withdrawn from the U.S.market in 2000 due to serious cardiac arrythmias [Wikipedia (2018);Hennessy,Leonard,Newcomb et al.(2008);Wysowski and Bacsanyi (1996)].It is consistent with our prediction result that cisapride has a high probability (prediction score=1.00) to cause ventricular arrhythmia.The above result further validated that our approach is not only capable to predict side-effects for a drug accurately and but also capable to find out reasons why a drug is withdrawn.

Table3:Validation examples of side-effects predicted for 10 uncharacterized drugs withdrawn from the market
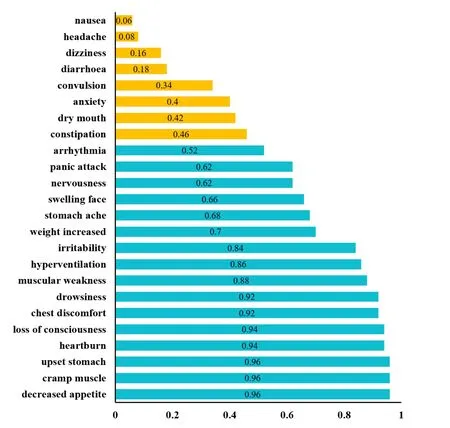
Figure4:Prediction scores of 24 side-effects which were mapped from DrugsCom into DrugBank with high credibility.The x-axis and y-axis represent the prediction scores and the side-effect names respectively
4 Conclusions
In this work,we proposed a novel method to predict potential drug side-effects based on drug chemical structures,drug interacted proteins and associated genes with sample selection and bipartite local models.The originality of the proposed method lies in the integration of three different drug features into a unified framework to measure similarities between drugs,in the development of balanced sample selection framework,in the implementation of bipartite local models.As far as we know,no existing work gathers all the above features in the field of drug side-effect prediction.In the performance evaluation,the proposed method showed the best performance on all evaluation metrics.The independent test on uncharacterized drugs demonstrate that the proposed method is practically useful in predicting both existing and new drug-sideeffect associations.
The proposed method is of value to various stages of drug development.At the early stage of drug candidate selection,the method could help to judge whether a compound should be chosen for further study or dropped due to unwanted side-effects.When the drugs are marketed,the method could help to find new indications for old drugs.This process is called drug repositioning,which could save a large amount of financial costs and time [Lee,Choi,Park et al.(2017)].Besides,warnings about potential serious side-effects can be given to the public before causing serious damages.The idea of balanced sample selection and bipartite local models also provides a new solution for unbalanced classification.
Additional files
The additional files for this work can be downloaded from:https://drive.google.com/open?id=0B9QA_8VX0i99S0NaZXpLOXUyYmc.
Acknowledgement:The work is supported by National Natural Science Foundation of China under Grant No.U1811462.
杂志排行

Computers Materials&Continua的其它文章
- A Capacity Improving Scheme in Multi-RSUs Deployed V2I
- A Multi-Objective Decision-Making Approach for the Optimal Location of Electric Vehicle Charging Facilities
- Multi-Rate Polling:Improve the Performance of Energy Harvesting Backscatter Wireless Networks
- A Novel Scene Text Recognition Method Based on Deep Learning
- Fuzzy C-Means Algorithm Automatically Determining Optimal Number of Clusters
- Ab Initio Molecular-Dynamics Simulation Liquid and Amorphous Al94-x Ni6 Lax (x=3-9) Alloys