基于转录组的大头金蝇密码子的偏好性分析
2019-07-22张玉波周正湘吴小玉
张玉波 周正湘 吴小玉

摘要:使用Codon W软件分析大头金蝇[Chrysomya megacephala(Fabricius,1794)]转录组10 923条全长转录序列的密码子偏好性。结果表明,大头金蝇转录组中的AT含量(61.81%)远大于GC含量(38.19%);PR2(parity rule 2,即密码子偏好性)绘图结果显示,密码子第3位碱基A的使用频率大于碱基T,碱基G的使用频率大于碱基C;中性绘图结果显示,该序列密码子的使用更多地受到选择压力的影响;有效密码子数(effective number of codons,简称ENC)在25.17~61.00个之间,均值为43.16个;密码子适应指数(codon adaptation index,简称CAI)在0.099~0.554之间,均值为0.215 8。结果共筛选出29个同义密码子相对使用度(relative synonymous codon usage,简称RSCU)>1的密码子和28个最优密码子。
关键词:大头金蝇;转录组;密码子偏好性;同义密码子
中图分类号: Q969.451.9;S186 文献标志码: A 文章编号:1002-1302(2019)11-0078-04
转录组测序(RNA sequencing)是指利用第二代高通量测序技术进行的cDNA测序,是一类专注于功能位点的测序策略,能全面快速地获取研究材料的特定组织在某一状态下的全部转录本信息[1]。随着高通量测序技术的应用[2],转录组测序以其较高的性价比而广受各位学者欢迎,被广泛应用于动植物的基因挖掘、功能鉴定等方面的研究,成为当前生物学研究的热点[3]。密码子偏好性指在编码氨基酸合成蛋白时,往往优先使用某一种或几种密码子[4],被优先选用的密码子称为最优密码子,这一现象广泛存在于生物类群中[5]。密码子偏好性具有物种特异性,不同基因组在进化过程中承受不同的选择压力,因此不同物种间密码子的使用偏好性不同[6-7]。分析密码子的偏好性可以深入了解编码序列的碱基含量、二核苷酸偏向性和隐藏的剪接信号等基因序列特征,这些都与密码子使用偏好性相关,都可以影响基因合成的设计与蛋白表达[7]。 大头金蝇[Chrysomya megacephala (Fabricius,1794)]为重要的卫生昆虫,隶属于丽蝇科(Calliphoridae)金蝇属(Chrysomya)[8]。研究大头金蝇转录组密码子偏好性,可以揭示氨基酸翻译过程中高表达与低表达基因对密码子的偏好选择,有助于解释其特殊生理效应的遗传机制,进一步为相关基因的克隆与表达奠定基础。
1 材料与方法
1.1 序列的获取
本研究数据来源于美国国立生物技术信息中心(National Center for Biotechnology Information,简称NCBI)网站,序列号为SRP050024,利用Codon W 1.4.2软件分析大头金蝇转录组10 923条序列的密码子偏好性。大头金蝇转录组测序数据见表1。
1.2 数据分析
1.2.1 碱基含量及PR2(parity rule 2,即密码子偏好性)的绘图分析 利用Codon W(version 1.4,http://bioweb.pasteur.fr/seqanal/ interfaces/codonw.html)软件对大头金蝇的转录组基因序列进行分析,计算每条序列的密码子总GC含量、第3位密码子的GC含量(GC3)、有效密码子数(effective number of codons,简称ENC或Nc)及密码子适应指数(codon adaptation index,简称CAI)。分别统计密码子3个位置上的GC含量,密码子第1位的GC含量表示为GC1,第2、3位的GC含量依次表示为GC2、GC3。GC1、GC2的平均值记为GC12。以GC12为纵坐标、GC3为横坐标进行中性绘图(neutrality plot),分析密码子第1、2位与第3位碱基组成的相关性,研究密码子偏性的影响因素。选择丝氨酸(TCA、TCC、TCG、TCT)、亮氨酸(CTA、CTC、CTG、CTT)、脯氨酸、精氨酸(CGA、CGC、CGG、CGT)、苏氨酸、缬氨酸、丙氨酸和甘氨酸,计算每个基因的A3/(A3+T3)和G3/(G3+C3),分析各基因密码子中4个碱基组分嘌呤(A和G)与嘧啶(T和C)之间的关系。
1.2.2 ENC及中性绘图分析 有效密码子数ENC用于检测单个基因密码子的使用偏好程度,取值范围在20~61之间,其值越低,表明该基因的密码子使用偏好性越强[9]。以密码子第3位上同义密码子GC的含量GC3s为横坐标、ENC为纵坐标,作二维散点图,探讨各基因密码子的使用偏性情况,并检测碱基组成对密码子偏性的影响。
1.2.3 同义密码子相对使用度及最优密码子分析 参照Sharp等的方法[10],同义密码子相对使用度(relative synonymous codon usage,简称RSCU)是对同义密码子使用偏好的评估[10],该值等于同义密码子的实际观测值与同义密码子平均使用期望值的比值。如果密码子的使用无偏好性,则RSCU值为1;如果该密码子比其他同义密码子的使用更频繁,则其RSCU值大于1,反之,RSCU值小于1。
利用高表达优越密码子分析方法[11],统计所有基因的ENC值、有序数据集上下10%区间内形成的高RSCU集合和低RSCU集合,進行最优密码子分析。根据2个子集的ΔRSCU值及卡方检验结果确定最优密码子。
2 结果与分析
2.1 碱基含量及PR2、中性绘图分析
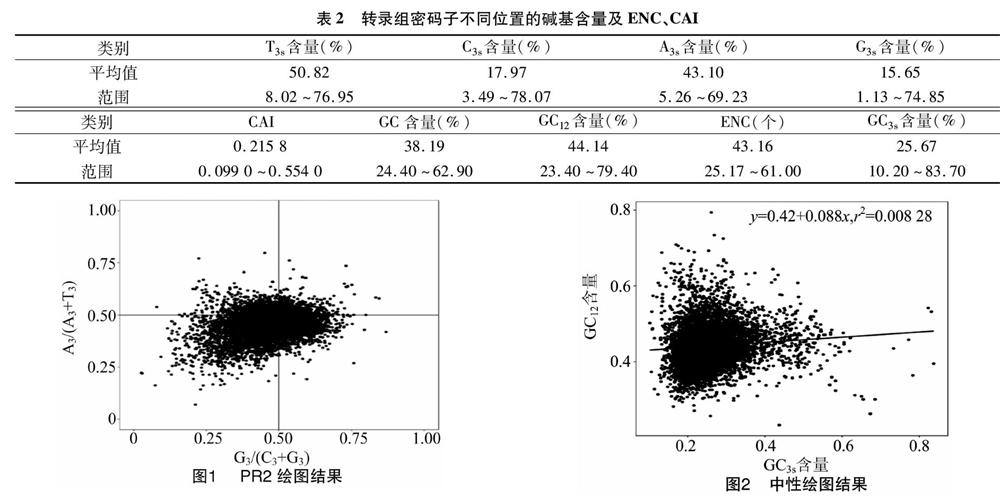
对已经得到的大头金蝇转录组数据进行筛选,共获得长度为300 bp以上的10 923条完全阅读框序列(全长CDS)。用Codon W软件进行密码子偏好性分析,结果表明,大头金蝇转录组序列中平均总GC量为38.19%,分布范围为 24.40%~62.90%,其中第3位点GC3s的平均值为25.67%,范围为10.20%~83.70%;总A、T、C、G 4种碱基含量分别为 32.9%、28.9%、18.4%、19.7%,密码子第3位点T3s、C3s、A3s和G3s含量的平均值分别为50.82%、17.97%、43.10%和 15.65%;GC12含量的均值为44.14%(23.40%~79.40%)(表2)。可以看出,在大头金蝇转录组序列中的AT碱基含量远高于GC。由图1可以看出,经PR2分析,大头金蝇转录组序列密码子第3位点碱基使用不均衡,密码子第3位碱基A的使用频率小于碱基T,碱基G的使用频率小于碱基C,表明大头金蝇转录组序列中基因密码子的使用模式受到突变压力和自然选择等多重因素的影响。
由图2的中性绘图结果可以看出,GC12含量与GC3含量呈现出负相关,但相关性不明显(r2=0.008 28),说明大头金蝇转录组序列的密码子受到的突变压力较小,GC含量较为保守,其密码子的使用更多地受到选择压力的影响[12-14]。
2.2 ENC、CAI的分析结果
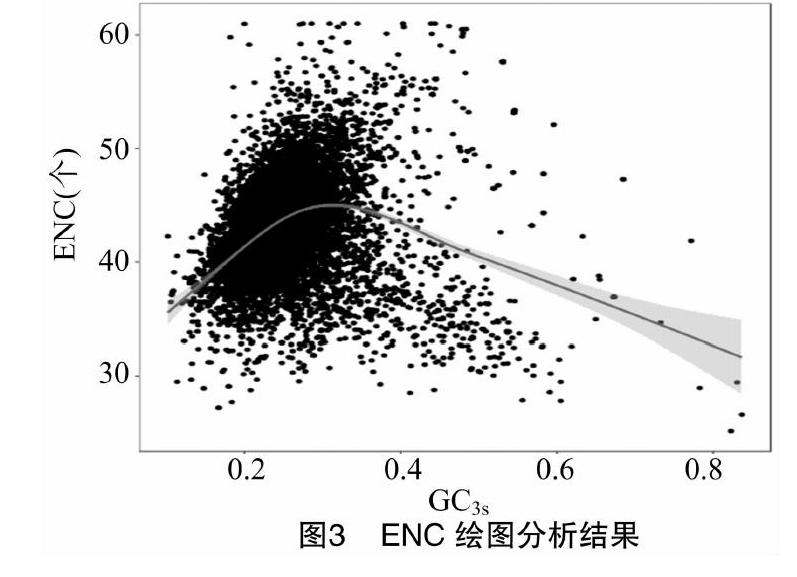
有效密码子数是衡量基因密码子偏好性的一个重要指标,数值范围为20个(每个氨基酸只使用1个同义密码子的极端偏好情况)~61个(每个同义密码子被平均使用的无偏好情况)。研究表明,当ENC≤35个时,基因密码子的使用偏好性随ENC值的降低而增强[15]。大头金蝇转录组序列的ENC在25.17~61.00个之间,均值为43.16个(表2),在 10 923 条序列中仅有359条序列的ENC小于35个[16]。CAI在0.099~0.554之间,均值为0.215 8。说明大头金蝇转录组中整体密码子偏好性较低,只有极少部分序列具有较强的
密码子偏好性。以ENC為纵坐标、GC3为横坐标进行ENC绘图分析发现,大部分序列沿标准曲线分布,小部分序列位于标准曲线以下较远的位置(图3), 说明大头金蝇转录组的密码子偏好性主要是受突变的影响,而选择压力仅在小部分序列中表现得比较明显。
2.3 同义密码子及最优密码子分析
经过计算可知,13条编码蛋白基因密码子中RSCU大于1的共有29个,除色氨酸(Trp)外,其他19种氨基酸及终止子均有RSCU值大于1的密码子。在这29个使用度较高的密码子中,第3位点嘌呤为U的有15个,为A的有12个,为G的有1个,为C的有1个,可以看出,在使用度较高的密码子中,绝大部分以A或U结尾(表3)。
采用ΔRSCU值法对大头金蝇转录组序列进行最优密码子的确定,共筛选出UUC、UUG、CUC、AUU、AUC、GUU、GUC、UAC、CAC、CAA、AAC、AAG、GAC、GAA、UCU、UCC、AGC、CCU、CCC、ACU、ACC、GCU、GCC、UGC、CGU、CGC、GGU、GGC共28个最优密码子,分别编码Phe、Leu、Ile、Val、Tyr、His、Gln、Asn、Lys、Asp、Glu、Ser、Pro、Thr、Ala、Cys、Arg、Gly共18种氨基酸(表4)。这28个最优密码子中以C结尾的有16个,以U结尾的有8个,以A、G结尾的均为2个,这与高频密码子的统计结果相似,说明大头金蝇最优密码子偏向于以C、U结尾。
3 讨论
目前已完成的双翅目类群转录组的测序工作不多,基于昆虫转录组的密码子偏好性分析结果更少,本研究结果与其他昆虫类群转录组密码子的使用模式是否一致,还需进一步分析确定。因此,若需要明确昆虫基因组密码子的使用模式及其与基因表达等之间的深入关系,则需要对昆虫线粒体基因组数据进行大量统计分析,而目前各数据库中有关昆虫线粒体基因组的数据相对较少,是否可以借鉴真菌、植物等真核生物线粒体成功测序的经验完成大量昆虫基因组的测序,进而为其密码子的真正“解密”提供原始材料,有待进一步研究。
参考文献:
[1]贾新平,孙晓波,邓衍明,等. 鸟巢蕨转录组高通量测序及分析[J]. 园艺学报,2014,41(11):2329-2341.
[2]Margulies M,Egholm M,Altman W E,et al. Genome sequencing in microfabricated high-density picolitre reactors[J]. Nature,2005,437(757):376-380.
[3]张棋麟,袁明龙. 基于新一代测序技术的昆虫转录组学研究进展[J]. 昆虫学报,2013,56(12):1489-1508.
[4]Olejniczak M,Uhlenbeck O C. tRNA residues that have coevolved with their anticodon to ensure uniform and accurate codon recognition[J]. Biochimie,2006,88(8):943-950.
[5]Campos J L,Zeng K,Parker D J,et al. Codon usage bias and effective population sizes on the X chromosome versus the autosomes in Drosophila melanogaster[J]. Molecular Biology and Evolution,2013,30(4):811-823.
[6]时 慧,王 玉,杨路成,等. 茶树抗寒调控转录因子ICE1密码子偏性分析[J]. 园艺学报,2012,39(7):1341-1352.
[7]Quax T E F,Claassens N J,Sll D,et al. Codon bias as a means to fine-tune gene expression[J]. Molecular Cell,2015,59(2):149-161.
[8]薛万琦,赵建铭. 中国蝇类(下册)[M]. 沈阳:辽宁科学技术出版社,1998:1438-1452.
[9]Wright F.The ‘effective number of codons used in a gene[J]. Gene,1990,87(1):23-29.
[10]Sharp P M,Li W H. The codon adaptation index-a measure of directional synonymous codon usage bias,and its potential applications[J]. Nucleic Acids Research,1987,15(3):1281-1295.
[11]Bellgard M,Schibeci D,Trifonov E,et al. Early detection of G+C differences in bacterial species inferred from the comparative analysis of the two completely sequenced Helicobacter pylori strains[J]. Journal of Molecular Evolution,2001,53(4/5):465-468.
[12]Sueoka N. Directional mutation pressure and neutral molecular evolution[J]. Proceedings of the National Academy of Sciences of the United States of America,1988,85(8):2653-2657.
[13]Sueoka N. Two aspects of DNA base composition:G+C content and translation-coupled deviation from intra-strand rule of A=T and G=C[J]. Journal of Molecular Evolution,1999,49(1):49-62.
[14]Nie X J,Deng P C,Feng K W,et al. Comparative analysis of codon usage patterns in chloroplast genomes of the Asteraceae family[J]. Plant Molecular Biology Reporter,2014,32(4):828-840.
[15]Comeron J M,Aguadé M. An evaluation of measures of synonymous codon usage bias[J]. Journal of Molecular Evolution,1998,47(3):268-274.
[16]Rai A,Yamazaki M,Takahashi H,et al. RNA-seq transcriptome analysis of Panax japonicus,and its comparison with other panax species to identify potential genes involved in the saponins biosynthesis[J]. Frontiers in Plant Science,2016,7:481.
