UPLC-MS/MS同时测定土壤中19种植物激素方法的建立和验证
2019-07-13卢玉秋宋阿琳唐治玉李艳玲董炜灵王恩召唐治喜范分良
卢玉秋,宋阿琳,唐治玉,李艳玲,董炜灵,3,王恩召,唐治喜,范分良*
(1 中国农业科学院农业资源与农业区划研究所/农业部植物营养与肥料重点实验室,北京 100081;2 中国计量科学研究院,北京100029;3 中南大学,长沙 410083)
植物激素是植物体内可自身合成的微量有机物,在非常低的浓度条件下就能参与并调控植物的生长、发育过程[1]。除植物外,微生物也能产生植物激素,如固氮螺菌[2]、土壤杆菌[3]、草生欧文氏杆菌[4]能够分泌细胞分裂素,赤霉菌[5]、巴西固氮螺菌[6]能分泌赤霉素,豆包菌[7]、固氮螺菌[2]、根瘤菌[8]、土壤杆菌[9]能够分泌生长素,这些微生物分泌的植物激素释放到土壤中,共同参与调控植物的生长发育。由于土壤基质成分复杂且土壤中植物激素含量极低,这在很大程度上增加了检测的困难,前人关于土壤中植物激素的提取和检测,大部分关注的只是一种或两种植物激素,土壤中多种植物激素的快速提取及同时检测鲜有报道,因此,土壤中植物激素的定性定量研究目前仍然是植物激素研究领域的难点之一。
植物中的激素最早使用生物鉴定法进行测定,它通过植物激素作用于植物组织或者器官后可产生生理生化变化,然后通过该变化的大小即可推算出植物激素的含量[10],如Went建立的燕麦鞘弯曲测定法测定生长素[11]、Thimann和Bonner建立的燕麦叶鞘切断伸长法测定生长素[12]等,生物测定法简单,但灵敏度和选择性较差[13];随后发展了免疫检测法[14],包括放射免疫分析和酶联免疫法,免疫检测法专一性强、选择性和灵敏度高[15],不过抗体容易发生交叉反应,且制备抗体时间长[16];之后发展了色谱法[17-18],该方法具有高分离特性,能够实现多种植物激素的同时测定,但对样品的前处理要求高,且对复杂基质样品定性定量存在误差[19];如今,色谱质谱联用技术[20-23]已经逐渐发展成为植物激素检测的主要方法[23],它结合了色谱具有对复杂样品的高分离能力和质谱拥有的高灵敏度、高选择性特点,大大提高了植物组织中激素检测的灵敏度[24],以及定性和定量的准确度[25]。相比之下,关于土壤中植物激素检测报道则少得多[26-28],目前检测的方法主要集中在电化学法[29-30]、比色法[31]、免疫检测法[27,32]和色谱法[33-35]。这些方法较色谱质谱联用技术相比,操作步骤复杂,灵敏度更低,而且对复杂样品基质的定性定量常常不如人意[19]。少量采用色谱质谱联用技术的研究,大多采用过夜浸提,但关注的植物激素种类单一,以往的研究没有成功的进行过土壤样品依次提取从而同时测定多种植物激素的方法,因此,有必要探索同时检测土壤中多种植物激素的方法,系统深入探明土壤植物激素的种类和含量。
本研究以中国科学院植物研究所试验地玉米根际土壤为研究对象,以超高效液相色谱-串联质谱法(UPLC-MS/MS)为研究手段,旨在建立和验证一种能同时测定土壤中生长素类、细胞分裂素类、赤霉素类、脱落酸类、水杨酸类、茉莉酸类、独脚金内酯、油菜素内酯8大类植物激素的方法。
1 材料与方法
1.1 仪器与试剂
1.1.1 仪器 Agilent1290超高效液相色谱仪、ABQtrap5500质谱仪,质谱仪配置有电喷雾电离接口及Analyst数据处理系统;超声波清洗仪;赛默飞RC10-22T真空离心浓缩机;Sigma3k15离心机;Mettler Toledo电子天平 (0.001 g);0.22 μm尼龙针头滤器。
1.1.2 试剂 甲酸为分析纯,异丙醇、二氯甲烷、乙腈、甲醇为色谱纯 (购自北京万诚博达科贸有限公司);实验室用水产自Milli-Qplus超纯水机;脱落酸(ABA)、赤霉素 (GA3)、茉莉酸 (JA)、吲哚-3-乙酸(IAA)、反式玉米素 (tZ)、玉米素核苷 (tZR)、异戊烯基腺嘌呤 (IPR)、吲哚-3-丁酸 (IBA)、N6-异戊烯基腺嘌呤 (IP)、独脚金内酯 (SL)、玉米素 (cZ)、二氢玉米素核苷 (DZR)、吲哚-3-丙酸 (IPA)、吲哚-3-乙酸甲酯(MeIAA)、水杨酸 (SA)、二氢玉米素 (DZ)、吲哚-3-羧酸 (ICA)、茉莉酸甲酯 (MeJA)、油菜素内酯 (BL)(购自 OlChemImLtd,Olomouc,Czech Republic)。
1.2 植物激素提取
供试土壤为中国科学院植物研究所试验地玉米根际土壤,其化学性质如下:有机质11.55 g/kg,全氮0.68 g/kg,全磷0.57 g/kg,硝态氮16.71 mg/kg,铵态氮6.99 mg/kg,速效磷42.78 mg/kg,有效钾65.64 mg/kg,pH为7.61。
提取步骤如下:1) 准确称取0.5 g (精确到0.001 g) 保存于-20℃冰箱的根际土壤样品于50 mL离心管;2) 加入5 mL异丙醇∶水∶甲酸(80∶19∶1,V/V/V);3) 在涡旋仪上涡旋30 s;4) 用超声波清洗仪于常温、100 W条件下超声30 min;5) 加入1 mL二氯甲烷,继续用超声波清洗仪于常温、100 W条件下超声30 min;6) 于9000 rpm、4℃的条件下离心10 min,将上清液转移至15 mL离心管;7) 在常温条件下用真空离心浓缩机对上清液进行离心浓缩;8) 用300 μL甲醇进行复溶;9) 复溶液过0.22 μm尼龙针头滤器,得到植物激素待测液;10) 采用UPLC-MS/MS法测定植物激素。
1.3 色谱-质谱条件
色谱条件:Waters ACQUITYUPLC ® HSS T3色谱柱 (2.1 mm × 150 mm,1.8 μm);分别以 0.30 mmol/L甲酸铵水溶液 (含0.01%甲酸) 作为流动相A和0.30 mmol/L甲酸铵乙腈 (含0.01%甲酸) 作为流动相B;进样量5 μL,流速0.30 mL/min,柱温30℃,梯度洗脱程序如表1所示。
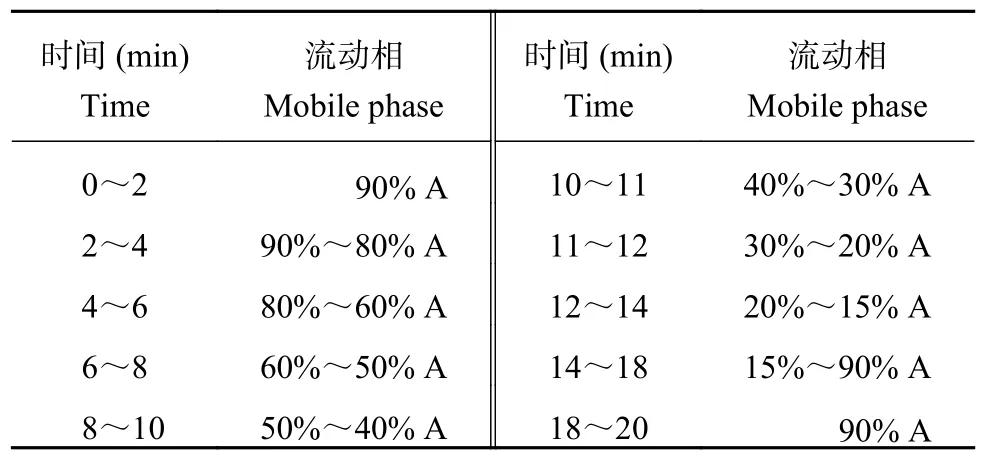
表1 梯度洗脱程序Table 1 Gradient elution procedure
质谱条件:离子源采用电喷雾离子源;扫描方式分别进行正/负离子模式扫描;检测方式为多反应离子监测 (MRM);气帘气压 (CUR) 20 Psi;碰撞气压 (CAD) medium;喷雾电压 (IS) 5500 V/-4500 V;离子源温度550℃;雾化气压 (GS1) 20 Psi;辅助气压 (GS2) 0 Psi;流速 0.30 mL/min。
1.4 定性与定量分析
对玉米根际土壤植物激素提取液进行浓缩、复溶、过滤后,采用UPLC-MS/MS法进行检测,以各植物激素标准品的定性离子对 (m/z) 及对应的的保留时间(RT)作为依据进行定性分析,以各植物激素标准品的定量离子对 (m/z) 所对应峰面积进行定量,并采用外标法计算回收率。测试设置5个重复,实验数据用Excel2010及Origin8.5软件进行分析。
2 结果与分析
2.1 色谱条件
本试验选择Waters ACQUITYUPLC ® HSS T3反相色谱柱来分离各植物激素,分别以0.30 mmol/L甲酸铵水溶液 (含0.01%甲酸) 作为流动相A和0.30 mmol/L甲酸铵乙腈 (含0.01%甲酸) 作为流动相B,最后采用上述1.3所述的梯度洗脱程序,实现了19种植物激素的完全分离。图1为19种植物激素标准溶液正、负离子扫描模式总离子流图(TIC),其中(a)、(b) 分别为正离子、负离子扫描模式TIC图。
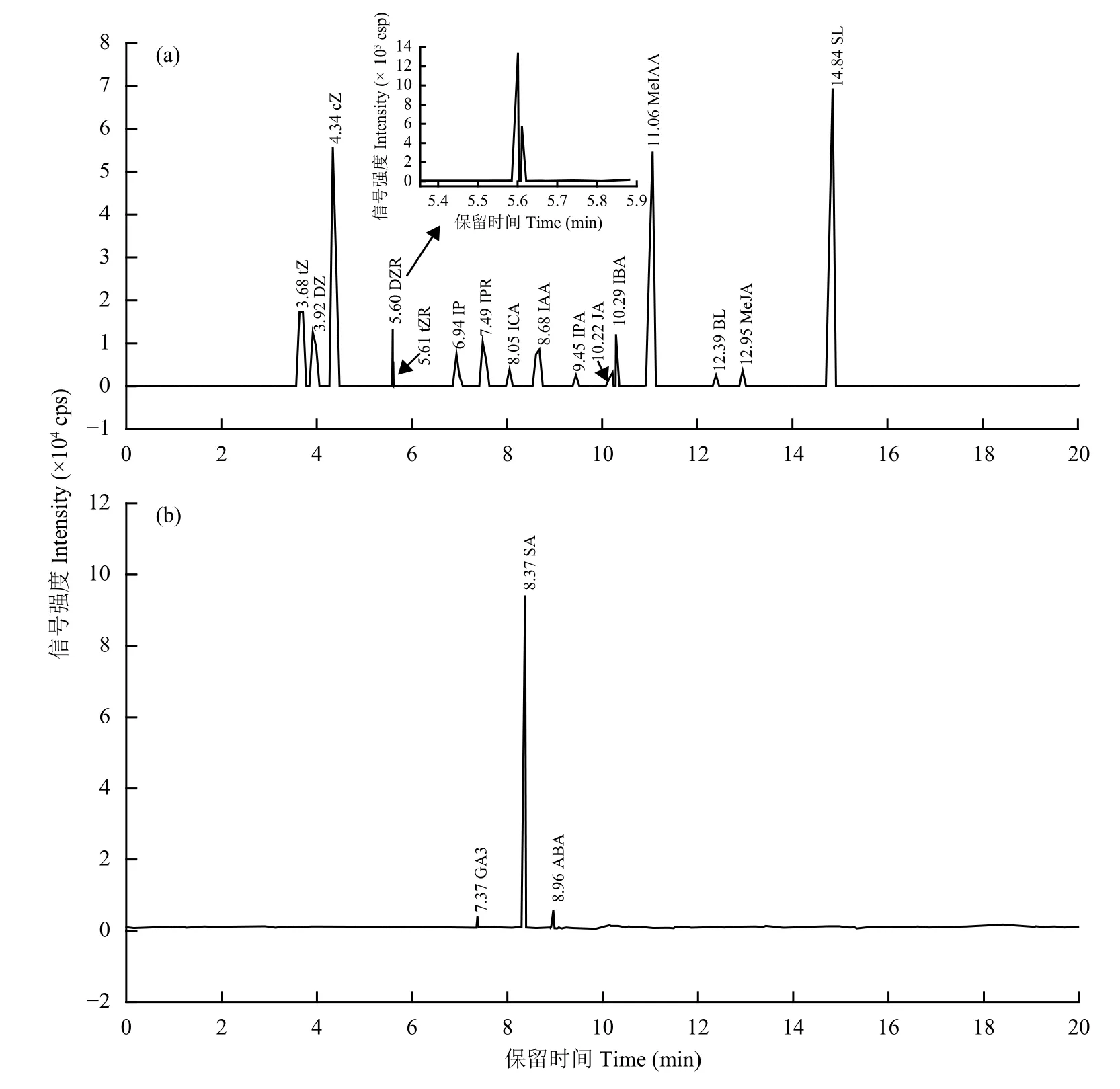
图1 19种植物激素标准溶液正离子 (a)、负离子 (b) 模式总离子流图Fig.1 Total ion chromatogram (TIC) of 19 phytohormones standard solutions with positive and negative ions scan mode
2.2 质谱条件
首先,配制浓度为300 ng/mL的单一标准溶液,采用流动注射的方式,以流速为7 μL/min将单一标准溶液注入离子源,在质荷比m/z 100~500范围内进行扫描,在电喷雾离子源 (ESI)下分别进行正离子模式 (+) 和负离子模式 (-) 全扫描,以确定各标准物质的分子离子峰和最佳的电离方式。结果表明,在电喷雾正离子扫描模式下,JA、IAA、tZ、tZR、IPR、IBA、IP、SL、cZ、DZR、IPA、MeIAA、DZ、ICA、MeJA、BL全扫描的分子离子[M + H]+较理想;在电喷雾负离子扫描模式下,ABA、GA3、SA全扫描的分子离子[M - H]-较理想。然后再进行子离子峰 (Product MS2) 扫描,不断的调节碰撞能量(CE),以确定各标准物质合适的子离子峰,最后进行多反应离子监测扫描 (MRM),分别对19种植物激素的去簇电压 (DP)、碰撞能量 (CE)、入口电压(EP)、碰撞室出口电压 (CXP) 进行优化,使信号达到最佳的响应值。19种植物激素标准物质优化后的质谱参数见表2。
2.3 方法的标准曲线、浓度范围、线性方程及检出限
首先取配制好浓度为500 ng/mL的混合标准溶液,依次稀释配制成系列浓度为0.2、0.5、1、5、10、20、50、100 ng/mL的混合标准溶液,采用UPLC-MS/MS进行检测,以植物激素浓度为x轴,所对应的峰面积为y轴,绘制标准曲线,并进行线性回归,得到回归方程和相关系数 (r),以信噪比(S/N) 为3确定化合物的检出限 (LOD)。结果表明,tZ、SL、cZ、MeIAA、DZ在0.2~100 ng/mL的浓度范围内线性良好,tZR、IPR、IBA、DZR在0.5~100 ng/mL浓度范围内线性良好,ABA、GA3、IAA、IP、IPA在1~100 ng/mL浓度范围内线性良好,SA、ICA、BL在5~100 ng/mL浓度范围内线性良好,JA、MeJA在10~100 ng/mL浓度范围内线性良好,r均大于0.99,LOD介于0.02~1.06 ng/g之间(表3)。

表2 19种植物激素优化后的质谱参数Table 2 Optimized mass spectrometric parameters of 19 phytohormones
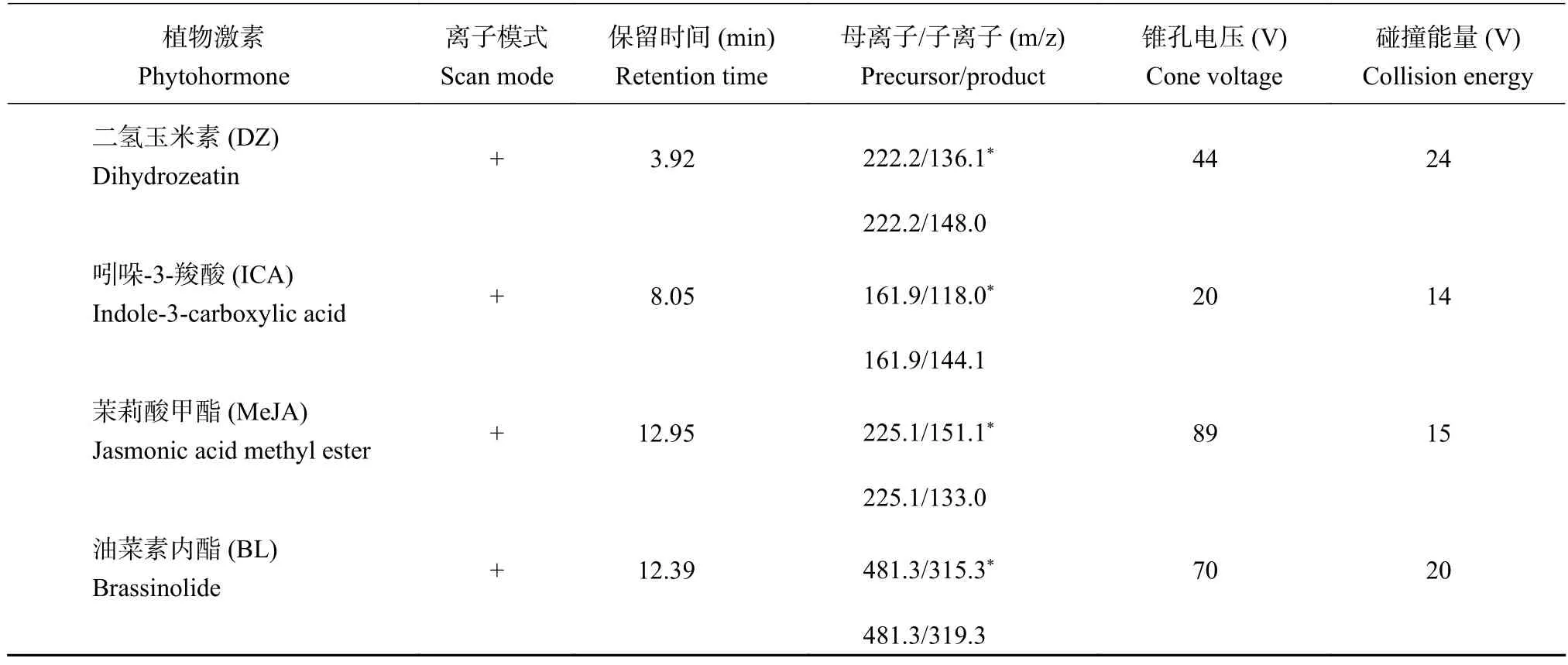
续表2 Table 2 continued
2.4 方法的回收率和精密度
为了考察实验方法的重现性以及准确度,分别向玉米根际土壤样品中添加低、中、高3种不同浓度的混合标准溶液,每个添加水平设置5个平行实验,并按照优化后的实验方法进行处理,最后通过计算方法的相对标准偏差(RSD)和加标回收率来衡量其精密度和准确度 (表4),表中未加标浓度为土壤基质样品中含有的植物激素。在3种不同浓度的添加水平下,玉米根际土壤中19种植物激素的回收率介于70.2%~117%之间,精密度介于0.2%~7.3%之间。
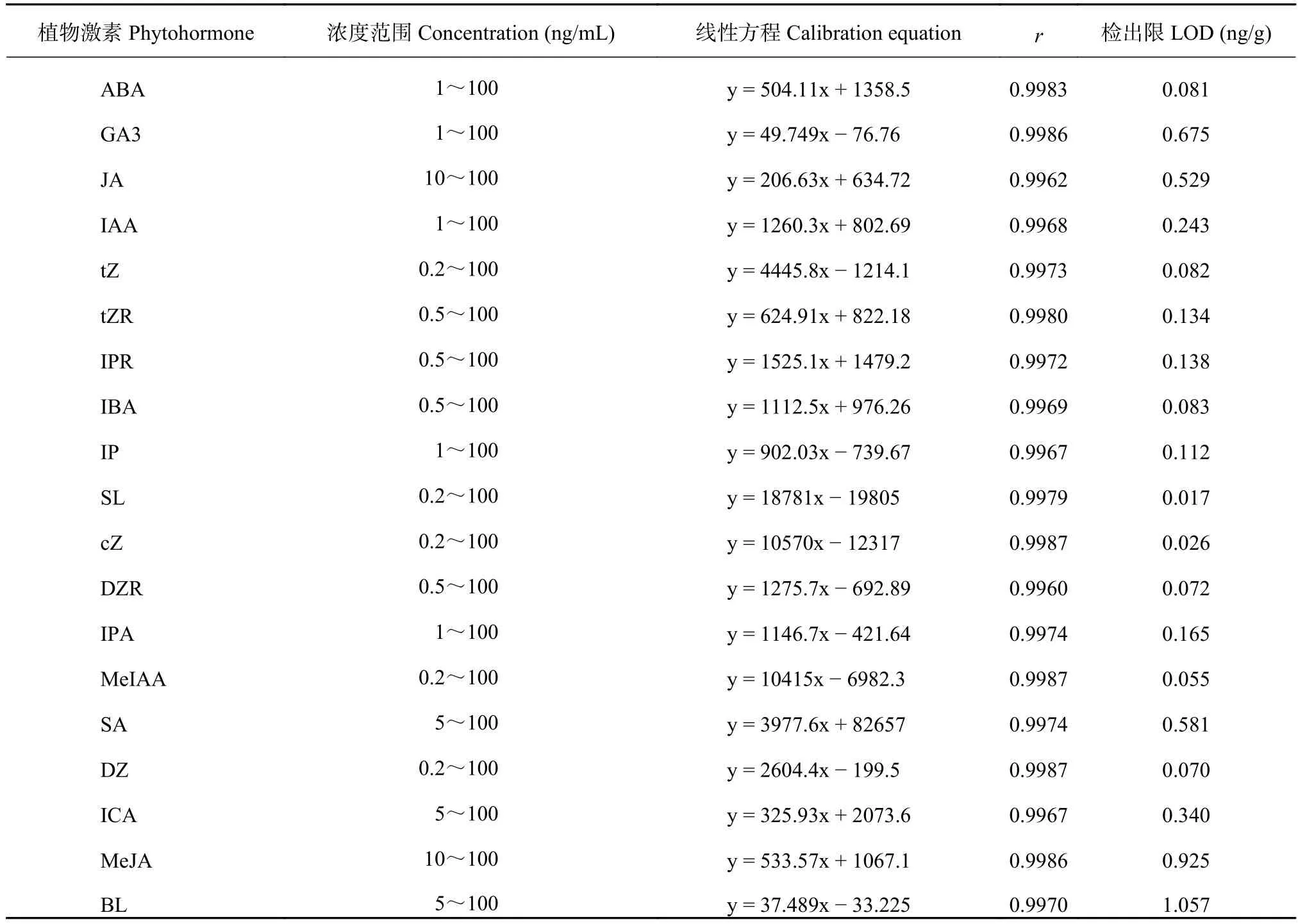
表3 19种植物激素的线性方程、相关系数及检测限Table 3 Linearity, correlation coefficient and detection limits of 19 phytohormones

表4 19种植物激素在土壤中的回收率和相对标准偏差 (n= 5)Table 4 Recovery rate and relative standard deviation of 19 phytohormones in soil
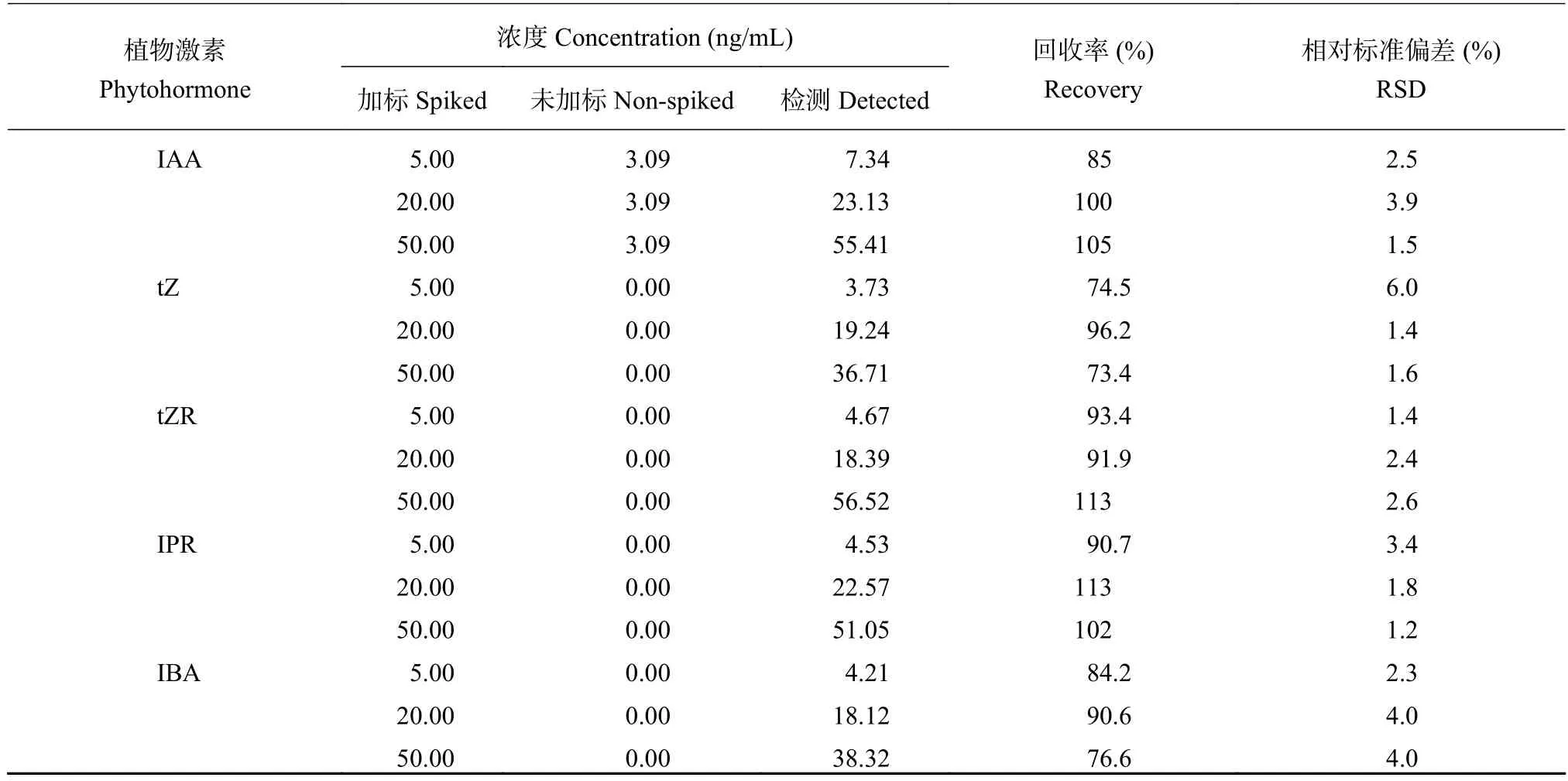
续表4 Table 4 continued
2.5 实际土壤样品测定
利用优化后的实验方法对玉米根际土壤植物激素进行测定,对同一份土样设置5个重复,测定结果表明:玉米根际土壤样品中检测出IAA、MeIAA、ICA、SL、SA 5种植物激素,含量分别为1.85 ±0.05、0.55 ± 0.00、5.79 ± 0.15、0.69 ± 0.00、3.94 ±0.50 ng/g。
3 讨论与结论
目前,关于植物激素的前处理方法,大多采用有机溶剂过夜浸提,重复浸提2~3次,并用固相微萃取小柱进行纯化,最后上机检测,这些方法前处理耗时长,采用固相微萃取小柱纯化虽然能够净化样品中的部分杂质,但是这种基于除杂的净化方法往往还会产生较大的基质效应[36],而且采用固相萃取小柱纯化不仅需要找到合适的清洗液以及洗脱液,此外活化、平衡、清洗、洗脱过程也很繁琐,损失也大,难以保证多种植物激素的回收率。本文的方法前处理不需要过夜浸提,而且加入二氯甲烷,离心浓缩复溶后不需要通过固相萃取柱即可直接进样分析检测,可以达到理想的分离效果,大大缩短了样品的前处理时间。
土壤中植物激素的检测方法从化学法[29-30]、酶联免疫法[28,32]、色谱法[33-35]到色谱质谱联用法[37],测试的灵敏度逐渐变高、专一性逐渐增强、准确度逐渐提高。超高效液相色谱-串联质谱法是近几年发展起来的测定植物激素的新方法[38],它结合了色谱的高分离性能及质谱的高灵敏度特性,大大提高了检测的灵敏度[24],可实现多种植物激素的同时检测。与气相色谱质谱联用相比,UPLC-MS/MS可避免样品分析中繁琐的衍生化过程[39]。与气相色谱及液相色谱法相比,UPLC-MS/MS能减少或消除色谱当前由于工作原理的不同可能存在的定性错误,提高植物激素定性及定量的可靠性[40]。与免疫法相比,UPLC-MS/MS可避免分析过程中抗体产生的交叉反应对结果产生的影响及放射性物质对实验人员身体造成的危害[16]。与化学法相比,UPLC-MS/MS灵敏度更高,选择性更强,能够检测出土壤中存在且浓度更低的植物激素。综上,超高效液相色谱-串联质谱法能为土壤中多种植物激素的同时检测提供一种更为简便、快速、准确的方法。
本研究建立了土壤中多种植物激素的提取及其超高效液相色谱串联质谱测定法,并测定了土壤中8大类植物激素,最后检测出IAA、ICA、MeIAA、SA、SL 5种植物激素,含量在0.55~5.79 ng/g之间,本方法前处理不需要过夜提取,大大缩短了样品的前处理时间,方法的检测限介于0.02~1.06 ng/g之间,回收率为70.2%~117%,精密度介于0.2%~7.3%之间,该操作简单、灵敏度高、选择性好,适用于土壤中多种植物激素的同时检测。
