DBT基因突变型儿童枫糖尿病
2019-07-10冯伟田青
冯伟, 田青
枫糖尿病(maple syrup urine disease,MSUD)是一种罕见的常染色体隐性疾病,最早由Menkes等[1]于1954年描述,全球18.5万人中约有1人患有该病,在某些民族和种族人群中发病率可能更高[2]。MSUD的特点是支链氨基酸α-酮酸脱氢酶复合体的先天缺乏,导致支链氨基酸(branched-chain amino acids,BCAAs)转氨形成的相应支链α-酮酸(branched-chain keto acids,BCKAs)不能氧化脱羧,从而造成体内BCAAs(包括亮氨酸、异亮氨酸及缬氨酸)和BCKAs的积累[3]。由于患者脑内BCAAs及相应支链酮酸代谢物的积累,会对脑组织产生神经毒性作用,引起一系列神经系统表现,如厌食症、呼吸暂停、刻板运动等[4]。本研究对1例MSUD患儿的临床资料进行分析,以提高此疾病的认识和诊疗水平。
1 病历资料
1.1 基本病史 患儿男,日龄11 d,因“吐沫伴纳差7 d”入院。患儿系第1胎第1产,胎龄39周顺产,产重3 200 g,无窒息史,生后Apgar评分9分,患儿父母适龄结婚,患儿父亲的外婆和患儿母亲的外婆是亲姐妹,否认家族类似疾病患者。生后母乳喂养,具体量不详,患儿于入院前7 d出现吐沫伴纳差,无发热、气促,无吐奶、呛奶,无嗜睡、抽搐等,尿液中有类似枫糖的气味,大便稀糊状。体格检查:体温36.2 ℃,心率118次/分,呼吸36次/分。反应可,面色欠红润,皮肤、巩膜无黄染,前囟平软,心、肺及腹部查体无异常,四肢肌张力不高。辅助检查:血常规、肝肾功能、电解质、大小便常规、心肌酶谱、TORCH、血氨、乳酸等化验均未见明显异常;血串联质谱:亮氨酸为3 281.50 μmol/L(正常值57.16~246.95 μmol/L),缬氨酸为613.28 μmol/L(正常值46.16~231.70 μmol/L);尿气相色谱示:4-羟基苯丙酮酸、2-酮-3-甲基戊酸、2-羟基异戊酸、2-酮-异己酸均不同程度升高。脑电图:中度异常脑电图;头颅MRI提示:脑内多发对称性异常信号,符合MSUD脑病改变,见图1。
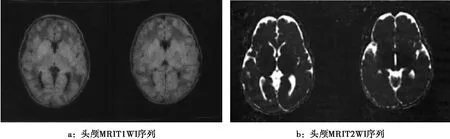
注:两侧大脑半球灰白质分界尚清楚,皮质板略薄,白质区域相对稍大,脑沟略浅,双侧中央回、放射冠、内囊后肢、丘脑区、小脑齿状核区见对称性长T2、DWI高信号影。
图1患儿生后14 d头颅MRI表现
1.2 基因测定 在取得知情同意后,采集患儿及其父母的外周血各2 mL,乙二胺四乙酸抗凝后送至北京金淮医学检验所利用安捷伦外显子芯片捕捉+高通量测序进行DX-09_支链氨基酸代谢障碍(枫糖尿症)检测。检测结果发现患儿父母均在chr1:100672078存在c.1132C>T(p.378X)的杂合突变(图2a),患儿chr1:100672078存在c.1132C>T(p.378X)的纯合突变(图2b)。提示该患儿在DBT基因发现一处恶性纯合突变,分别来自父母,符合美国医学遗传学与基因组学学会相关指南的Pathogenic级。

图2 基因测定结果
1.3 治疗及随访 患者入院后给予心电监护,静脉补液和肠外营养支持等对症治疗。在初步确诊为“MUSD”后,立即给予特殊配方奶粉以及维生素B1控制饮食治疗,患儿好转出院。出院后随访2.5年,现患儿精神食欲可,无特殊不适。测亮氨酸及缬氨酸水平分别为782.19 μmol/L、202.32 μmol/L,复查头颅MRI示:颅内多发异常信号范围较前明显变小,双侧脑室三角区脉络丛对称性异常信号灶(图3)。

注:双侧大脑半球灰白质分界清楚,原双侧中央回、放射冠、内囊后肢、丘脑区、小脑齿状核区见对称性长T2、DWI高信号影较前明显变小,其中内囊异常信号基本消失,双侧侧脑室三角区见对称性类圆形长T1长T2异常信号影。
图3患儿2岁6个月头颅MRI表现
2 讨论
目前大多数研究根据支链氨基酸α-酮酸脱氢酶活性及临床表现将MSUD分为经典型、中间型、间歇型、维生素B1有效型及脂酰胺脱氢酶缺乏型等5种类型,其中经典型是最严重、常见的表现型,新生儿早期即可发病,预后差,可迅速发展为呼吸衰竭、昏迷以致死亡[5]。MSUD临床表现缺乏特异性,早期常出现纳差拒奶、嗜睡、枫糖尿味等,后期进展为抽搐、惊厥、呼吸暂停、下肢交叉步等,如未得到及时干预可发生昏迷、中枢性呼吸衰竭,严重者可致死亡[4,6]。MSUD的标准治疗主要为特殊饮食及维生素B1的摄入,重症者则需要透析、血浆置换等积极处理。特殊饮食在恢复和维持MSUD患者的代谢稳态至关重要,应避免摄入含BCAAs的蛋白质、液体等,其目的是防止内源性BCAAs和BCKAs的合成和积累[7]。维生素B1是支链氨基酸α-酮酸脱氢酶的辅助因子,对于MSUD患儿,补充维生素B1可增加对饮食中BCAAs耐受性或降低血浆BCAAs,尤其是DBT突变患者[7-8]。本病例生后5 d出现纳差,生后11 d就诊,临床表现轻,及时予以特殊配方奶粉及维生素B1饮食治疗后患儿症状明显好转,随访BCAAs水平及头颅MRI均提示明显好转,根据发病时间、病情进展及维生素B1治疗有效,可把此病例考虑为维生素B1有效型。
MSUD还可根据BCKDHA、BCKDHB、DBT及DLD基因突变的不同将该病分为ⅠA型、ⅠB型、Ⅱ型、Ⅲ型4种基因型,其中以BCKDHA、BCKDHB点突变最为常见,DLD基因突变报道较少。在人类基因突变数据库(HGMD)中,已有88例BCKDHA、85例BCKDHB、71例DBT和21例DLD突变报道[9]。Mitsubuchi等[10]认为ⅠA型、ⅠB型倾向于表现为经典型,Ⅱ型患者临床表现较轻,且所有维生素B1有效型均是Ⅱ型。本患儿为DBT基因突变、临床表现轻、及时维生素B1治疗后明显好转,这符合Mitsubuchi等的见解。该病例中患儿父母均携带c.1132C>T(p.378X)的杂合突变,患儿DBT基因c.1132C>T(p.378X)的纯合突变,此突变位点还尚未见报道。且患儿父亲的外婆和患儿母亲的外婆是亲姐妹,笔者测该患儿DBT基因c.1132C>T突变具有家族性,蒋丰智等[11]也报道了1例临床症状轻、治疗后好转的家族性MSUD病例,疾病发展和该患儿相似,但本文患儿父母否认家族中有类似疾病患者,这为今后继续探索此类疾病提供了新的思考。
由于MSUD为支链氨基酸代谢障碍性疾病,体内相关BCAAs和BCKAs在血液及颅内积累,而且BCKAs可从尿液中排出,所以血串联质谱、头颅影像学及尿气相色谱筛查对疾病的早期诊断有重要意义。血串联质谱可发现亮氨酸、异亮氨酸及缬氨酸升高,亮氨酸升高更为显著,Schadewaldt等[12]提出血浆中异亮氨酸升高对于诊断MSUD更具有特异性,但异亮氨酸的测定在临床上尚未普及。头颅影像学:在急性期由于BCAAs及BCKAs的蓄积影响三羧酸循环,从而干扰脑细胞的能量代谢,导致Na+/K+ATP泵功能紊乱而弥漫性脑水肿表现[13]。慢性期由于BCAAs和BCKAs累积使脑白质逐渐发生海绵状变性和髓鞘形成障碍,可见对称性中央回、基底节、丘脑、齿状核、大脑脚等部位损害[14]。血串联质谱筛查可在MSUD患者出现脑病症状或头颅影像学改变前检测到血液中支链氨基酸的升高,因为脑内BCAAs及BCKAs积累对脑组织产生神经毒性作用,导致脑白质病变需要发展过程[15]。尿气相色谱可检测尿样中出现异常有机酸。本研究患儿在入院后即对其进行血串联质谱、尿气相色谱及头颅MRI筛查,其化验及检查结果均符合MUSD表现,加之发现尿有特殊枫糖味,初步诊断此病不难,这对MSUD进行及时干预改善预后极其重要。
早期诊断和早期干预可明显改善MUSD预后,所以对疑似MSUD患儿及时进行血串联质谱、尿气相色谱及头颅影像学筛查至关重要。MUSD特殊饮食及维生素B1治疗是改善症状、延缓病情发展的有效手段,目前已有报道肝移植术治疗MSUD,术后患者的支链氨基酸α-酮酸脱氢酶活性显著提高,支链氨基酸代谢改善[16],对生存质量和神经智力改善都是有益的。
