UHPLC-Q-TOF MS/MS测定稻米代谢物的方法研究
2019-07-08肖然,马莺
肖 然,马 莺
(哈尔滨工业大学化工与化学学院,黑龙江 哈尔滨 150090)
0 前言
水稻作为最重要的粮食作物,其代谢物尤其是次级代谢物不仅为植株生长提供能量,也是人类和动物营养物质的主要来源。次级代谢产物主要是指植物细胞代谢和调控过程的内源性小分子终产物。次级代谢物水平是生物系统对环境变化和遗传修饰的终端响应[1]。植物体中目前鉴定出的次级代谢物有20万~106万种,它们在植物的生长发育、抵御生物胁迫和非生物胁迫等方面发挥重要作用[2]。
植物产生的代谢物的种类多样性和含量差异性远远超过微生物乃至动物,因此对这些代谢物分析通常需要高灵敏度、高分辨、高通量的高效分析平台。核磁共振技术(Nuclear Magnetic Resonance,NMR)是一种高通量且无偏向性的分析手段,适用于少量代谢物的实时动态检测和结构鉴定,但NMR检测范围小,灵敏度较低,不适用于大范围植物次级代谢物的检测。气相色谱质谱联用法(Gas Chromatography-Mass Spectrometry,GC-MS)是最早应用于大规模植物代谢物分离研究的技术手段,它具有高灵敏度和高分辨能力,并且检测出的化合物可利用质谱数据库进行检索和比对。但GC-MS不适合分析热不稳定、不易挥发、分子量较大及极性较强的代谢物,如糖类、氨基酸等成分,需要衍生化才能得到较多的代谢物组分信息[3]。液相色谱质谱联用技术(Liquid Chromatography-Mass Spectrometry,LC-MS)不受待测物挥发性和热稳定性的影响,适合分离热不稳定、不易挥发及分子量较大的代谢物,尤其对生物碱、皂苷、酚酸及多胺等植物次生代谢物有较好的分析能力[4],LC-MS样品前处理较为简单、检测温度低、物质分离快速、高效,与MS高灵敏度、高专属性的优点结合,具有提纯和检测单一物质的能力,是目前是植物次级代谢产物相关研究中最常用的分析技术[5]。超高效液相色谱(UHPLC)比HPLC具有更快速、高效、灵敏的特点,与质谱联用时可减少基质干扰效应,从而提高质谱的检测灵敏度。近年来UHPLC-MS技术为植物次级代谢物研究提供了巨大的推动力[6-8]。
本实验旨在建立一种利用UHPLC-Q-TOF MS/MS方法对大米代谢物的分析方法,以稻米为研究对象,以识别出的特征峰个数和得到的总峰面积为评价指标,通过单因素实验分别考察样品前处理方式(包括提取溶剂,料液比,提取方式以及提取时间)对水稻种子中代谢物的提取效率及液相条件(包括梯度洗脱条件和色谱柱温)对样品分离效果的影响。
1 实验材料与方法
1.1 仪器与试剂
KQ-700D型数控超声波仪(昆山市超声仪器有限公司),3K15型高速台式冷冻离心机(德国Sigma公司),MX-S型涡旋仪(大龙兴创实验仪器(北京)有限公司),1290型UHPLC液相色谱系统(德国Agilent公司),6540 UHD型Q-TOF质谱仪(德国Aglient公司)。
乙腈(美国Merck公司),甲酸(美国Sigma有限责任公司)均为色谱纯,其他试剂均为分析纯。
1.2 试验方法
1.2.1 样品预处理
在样品制备前,水稻样本储存在干燥的冷藏室(-20℃,RH15%)。稻谷先进行脱壳和分离,再挑选出种粒饱满水稻颗粒,加入液氮进行研磨,并过80目筛,得到均匀的大米粉。样品储存于干燥的冷藏室中(-20℃,RH15%),避免阳光直射直至样品提取。
1.2.2 样品提取
称取600 mg的研磨过的大米粉末置于玻璃试管中,按不同料液比(1∶3,1∶5,1∶10,1∶20)加入三种提取溶液甲醇、乙腈及水(甲醇∶乙腈∶水=1∶0∶0,0∶1∶0,1∶1∶0,1∶0∶1,0∶1∶1,1∶1∶1)。涡旋混合器混匀10 s后,25℃条件下经不同提取时间(5 min,15 min,30 min,60 min,90 min)和三种提取方法(震荡,超声和涡旋),提取结束后涡旋混合器混匀10 s。提取混合物在4℃,8500×g条件下离心20 min。上清液转移至新的离心管中,过0.22 μm聚四氟乙烯膜滤器,收集滤液并存储在-80℃直至代谢物分析。以UHPLC-Q-TOF MS/MS结果所得特征峰个数和总峰面积为评价指标,对提取方法进行评价。
1.2.3 UHPLC-Q-TOF MS/MS分析
采用安捷伦1290 UHPLC色谱系统串联6540 UHD accurate-mass Q-TOF质谱仪对大米提取物进行分析。液相A液为0.1%(v/v)甲酸水溶液,B液为0.1%(v/v)甲酸乙腈水溶液。上样前,色谱柱在85%A液中平衡。在分析过程中,样品盘保持在4℃。分析时,样品以随机序列被注入到Acquity BEH C18色谱柱(2.1×150 mm,1.7 μm),上样量5 μL。质量控制(QC)样品每5次注射一次用于分析控制。在梯度洗脱中,流动相的流速设置为0.3 mL/min。质谱分析条件为:一级质谱采用射流电喷雾电离(Electrosprayionization,ESI)源,检测方式:正离子模式,脱溶剂气温度:325℃,流速:9 L/min,扫描时间:0.5 s,扫描范围:m/z 50-2000,雾化器压力:45 psi,毛细管电压:4000 V,进样锥锥孔电压:140 V,锥孔补偿电压:65 V,母离子累积时间,0.02 s/spectra;二级质谱采用信息依赖型扫描(information dependent acquisition,IDA),具体设置为Exclude isotopes within 4 Da,Candidate ion to monitor per cycle:6,high sensitivity模式,去簇电压:60 V,碰撞能量:35±15 eV,子离子累积时间,0.05 s/spectra,使用质量校正离子对(m/z 301.998139和1033.988109)对系统进行校正。
实验首先以获得色谱峰个数及总峰面积为评价指标,对三种液相梯度洗脱方法(10min,23min,30min)进行了研究,在确定梯度洗脱方法的条件下,进而以色谱峰个数及出峰时间对三种色谱柱温(25℃,30℃,40℃)进行考查。
表1液相色谱流动相梯度洗脱的方法
Tab.1 Gradient elution of UHPLC system
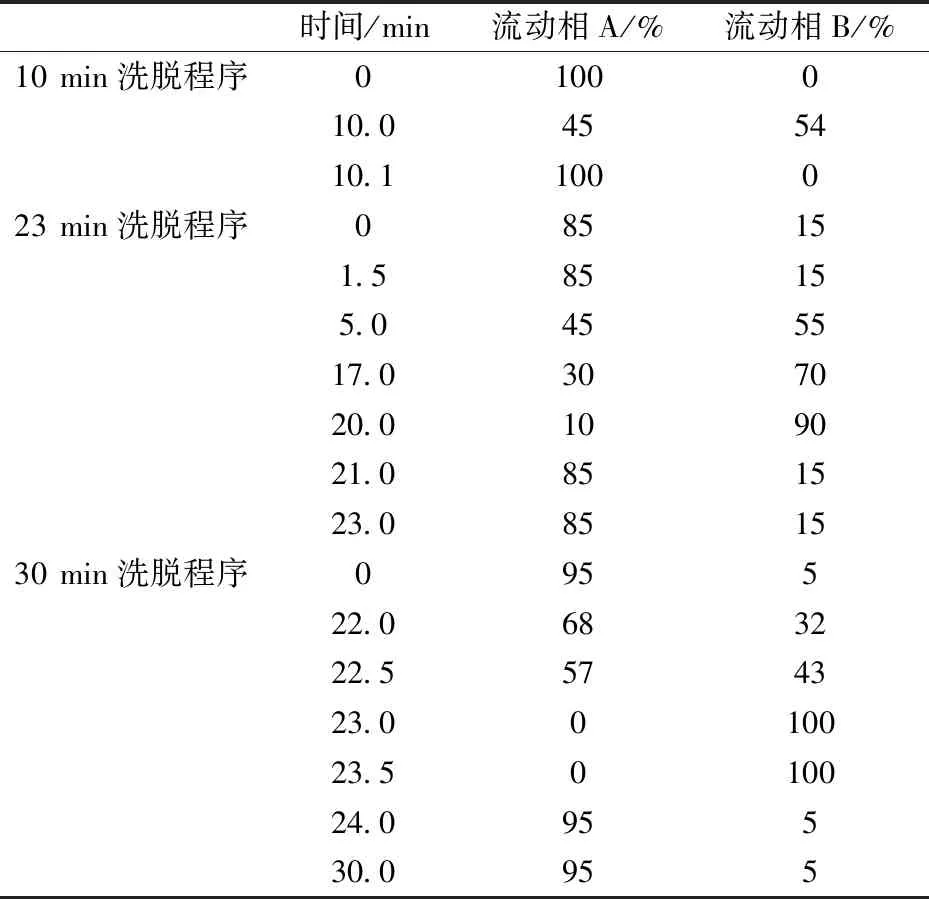
时间/min流动相A/%流动相B/%10min洗脱程序0100010.0455410.1100023min洗脱程序085151.585155.0455517.0307020.0109021.0851523.0851530min洗脱程序095522.0683222.5574323.0010023.5010024.095530.0955
1.2.4 数据处理分析
对得到的大米样品的UHPLC-Q-TOF MS/MS原始数据利用MS Convert软件将初始数据文件转换为mzML格式。并使用在R软件包(Ver.3.4.0)中运行的XCMS软件进行预处理,包括对总离子流色谱原始数据中的色谱峰的提取,峰对齐,去噪音等处理,得到各个峰的保留时间、峰高、去噪音等处理,得到各个峰的保留时间、峰高、峰面积和质荷比数据。数据处理后,生成由保留时间(RT)和质量荷电比(m/z)数据对组成的二维矩阵。
1.2.5 UHPLC-MS/MS方法的评价
重现性:按最佳前处理方法平行处理6份QC样品,连续测定6次,数据预处理后,统计各代谢物保留时间和响应值的相对标准偏差(Relative Standard Deviation,RSD)分布,考察方法重现性。
精密度:实验同时考察了UHPLC-Q-TOF MS/MS方法的日内精密度和日间精密度。日内精密度的测定:取制备好的同一QC样品分别在制备后的24小时内的0,4 h,8 h,12 h和24 h时间点进样,每次样品平行检测6次,统计各特征峰保留时间和响应值的RSD分布,考察方法的日内精密度。日间精密度的测定:取制备好的同一QC样品分别在制备后的24 h,36 h和48 h时间点进样,每次样品平行检测6次,统计各特征峰保留时间和响应值的RSD分布,考察方法的日间精密度。
线性范围:分别称取QC样品20,40,60,80,100,120,140和160 mg,按所确定的最佳前处理方法处理,每个样品量平行处理3次。数据预处理后,统计各共有特征峰响应值与样品量间的线性相关系数(r)的分布,考察方法的线性范围。
2 实验结果与讨论
2.1 样品提取方法的优化
稻米作为植物学样本,其代谢物种类繁多,且含量和极性差别较大,例如极性较强的糖、氨基酸类等,以及极性较弱的脂类、固醇类等代谢物[9]。不同的提取方法对样品后期分析会产生极大的影响,本实验采用不同提取试剂,料液比,提取方法及提取时间对稻米样品进行提取,通过特征峰个数和总峰面积考察不同溶剂对稻米代谢物整体提取效率和稳定性的影响。图1显示了不同条件对大米代谢物提取效率的影响。

(a.提取溶剂;b.提取料液比;c.提取方式;d.提取时间)(a.Material to liquid ratio;b.extraction solvent;c.extraction method;d.extraction time)图1 不同提取条件对大米代谢物提取效率的影响Fig.1 Effects of different conditions on extraction efficiency of rice metabolites
实验同时比较了不同配比的常用的三种提取溶剂,甲醇、乙腈和水对大米代谢物的提取效率。结果显示(图1a),50%的甲醇提取到的特征峰个数(约9 348个)和总面积均最大,其次是甲醇-乙腈-水(1∶1∶1)和100%甲醇,100%乙腈和50%乙腈提取效果最差,仅有5 442和5 988个特征峰被检测到,这可能是由于稻米当中糖类及其衍生物、氨基酸、有机酸等极性物质较多,所以极性较强的甲醇溶液可以获得更好的提取效果。适当的料液比可以显著增强大米代谢物的提取效率。如图1b所示,料液比为1∶3时,原料由于无法被充分浸润而导致提取效率过低(仅有3 792个特征峰被提取);当料液比大于1∶5后,提取出的代谢物总数增加并不显著,但代谢物总面积却因为溶剂过多以致代谢物浓度降低而降低。因此料液比1∶5被选定为后续实验的料液比条件。图1c显示了辅助超声、恒温振荡、涡旋3种常见的提取方法对水稻中物质提取效率的影响。其中辅助超声和恒温振荡提取时间为30 min,涡旋提取则选择30 min,每5 min间歇1 min的方式。由图1c可以看出,超声提取方法得到的代谢物个数(8 664个特征峰)和总峰面积(约7.88×106)均优于其他两种方法,且超声提取操作简单,所以选择超声提取方式进行进一步的筛选实验。在对提取时间的筛选实验中发现,较短的提取时间通常会保护代谢物的结构不受到提取过程的损坏(温度过高及机械损害),但过短的时间(5 min)也导致提取不完全的现象发生,因此实验选择30 min作为后续实验的提取时间。
2.2 UHPLC条件的优化
2.2.1 UHPLC梯度洗脱条件的优化
如何将经预处理的样品快速而稳定的分离和检测是UHPLC-MS/MS技术的关键。流动相梯度的变化能引起流动相极性的变化,从而调节样品中各组分在流动相中的分配系数(K值),以达到各组分良好的洗脱和分离的目的[10]。本实验以液相色谱所得色谱峰数量和总色谱峰面积为指标,对三种梯度洗脱条件(10 min,23 min,30 min)对UHPLC-Q-TOF MS/MS的检测能力的影响进行考察。
表2不同流动相梯度洗脱效果的评价
Tab.2 Evaluation of different gradient elution

梯度洗脱条件/min色谱峰数量/个色谱峰面积/×107108421.19±0.052319014.21±0.193024791.69±0.03
流动相梯度的变化导致化合物保留属性的改变,根据化合物相似相溶原理,极性的改变将改变化合物在固定相和流动相间的分配系数,达到洗脱的目的[11]。如表2所示,流动相极性的改变可以改变化合物的出峰个数,10 min对样品的洗脱能力较低,仅有842个色谱峰被分离出来;而30 min的洗脱则分理出2 479个色谱峰;但从色谱峰面积来看,30 min洗脱条件虽获得了较多的色谱峰数量,但总峰面积较低,仅为1.69±0.03×107,而23 min梯度洗脱技能获得相对较多的色谱峰数量(1 901个),还可兼顾得到较高的色谱峰面积(4.21±0.19×107)。因此在本研究中,23分钟梯度洗脱是最佳的流动相梯度,在该条件下,化合物得到有效的分离,同时检测容量也最大。
2.2.2 UHPLC色谱柱柱温的优化
通常情况下,色谱柱的柱温被控制在室温,然而对于大批量样品而言,长时间的UHPLC-MS/MS分析会导致柱温的变化,进而引发化合物的保留属性和色谱柱的柱效的变化[12]。本实验在确定流动洗脱条件的前提下,以液相色谱所得色谱峰数量,出峰时间和总色谱峰面积为指标,对三种色谱柱温(25℃,30℃和40℃)对UHPLC-Q-TOF MS/MS检测能力的影响进行考察。
柱温是高效液相色谱中比较重要但又常被忽略的因素。本研究我们考察了UHPLC-Q-TOF MS/MS系统在25℃,30℃和40℃三个不同温度下检测能力的差异,结果表明:柱温的改变导致化合物保留属性的改变,这主要是因为温度的升高促进了化合物从固定相中解吸的过程。如图2所示,特征峰1(红色箭头)相对保留时间为7.13 min,而当柱温升高至40℃时,相对保留时间变成了5.98 min,整个出峰时间缩短了1.15 min。从整体洗脱程序上来看,25℃时,主要色谱峰约在21 min出峰完毕,当柱温上升到40℃时,主要色谱峰出峰结束时间提前至18 min左右,整体出峰时间缩短约3 min。由此说明,升高柱温,可以加快化合物的出峰时间而缩短分析时间,减少流动相的使用,节约分析成本。一般而言,柱温越高,分离效率越高,分析时间越短,还可以降低柱压,改善峰形等[12]。但是高温一方面造成色谱柱填料的降解,减少色谱柱的使用寿命,另一方面容易引起色谱峰的分离程度降低,甚至造成双峰和平峰现象(红色虚线椭圆)。实验同时考察了色谱柱温对色谱峰个数的影响和总峰面积的影响。
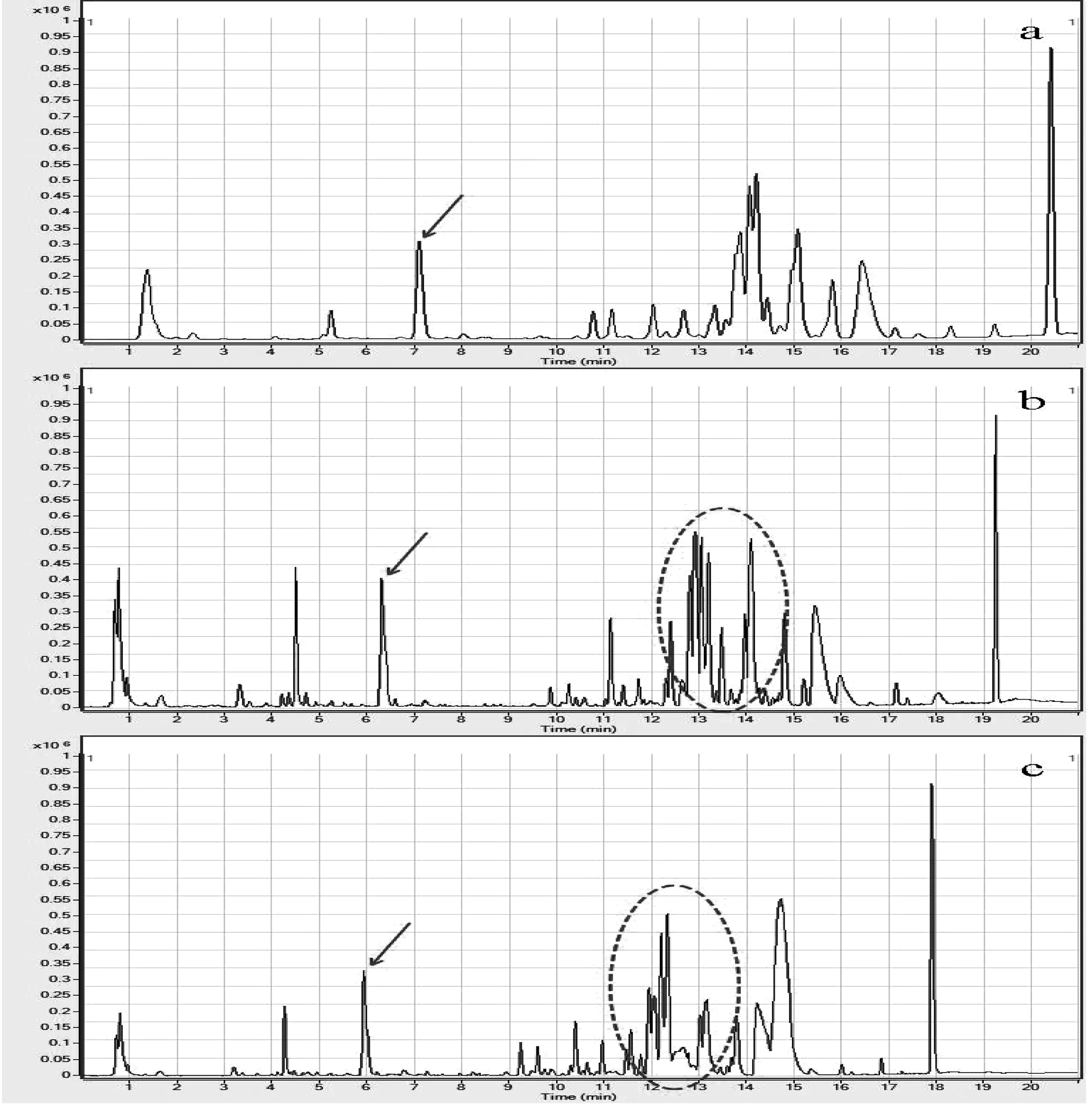
(a.25℃,b.30℃,c.40℃)(a.25℃,b.30℃,c.40℃)图2 液相色谱色谱柱温的优化Fig.2 Temperature optimization of chromatographic column applied in LC-MS/MS analysis
表3不同柱温对液相色谱影响的评价
Tab.3 Evaluation of different column temperatures

柱温条件/℃色谱峰数量/个色谱峰面积/×1072517993.97±0.233018214.52±0.074017674.12±0.62
由表3可以看出,色谱柱温的变化会引起色谱出峰数量和色谱峰面积的变化,随着柱温从25℃上升至30℃,可识别的色谱峰数量由1 799个增加至1 821个,然而当柱温从30℃上升至40℃后,色谱峰数量下降至1 767个,这与图2结果吻合,图2b中12~16 min的色谱峰,在升高柱温后发生了部分色谱峰的聚集及平峰现象(图2c中11~15 min)。色谱峰分离度的下降会引起质谱检测的误差增大,因此本实验中色谱柱温控制在30℃。
2.3 UHPLC-Q-TOF MS/MS方法的评价
为了确定上述研究中建立的UHPLC-Q-TOF MS/MS测定稻米代谢物的方法是否符合实际检测需要,实验以色谱峰的响应值的RSD和相关系数r为指标,从重复性,日内、日间精密度及线性范围四个方面对方法进行评价。
实验对重复性的考察结果(图3a)表明,有83.09%特征峰峰面积RSD小于20%,其峰面积累积响应值占全部特征峰的78.82%,峰面积RSD小于30%的占总个数的91.78%,其峰面积累积响应值占全部代谢物的88.65%以上。保留时间的RSD值仅为2.32%,由此可看出该方法的重复性较好。
通过连续3天对QC样品进行检测,并计算特征峰保留时间和峰面积RSD进行日内、日间精密度分析发现,保留时间的日内和日间RSD值均在5%以内(3.2%和4.7%);日内QC样本特征峰面积的RSD百分比在30%以内的有91.98%(图3b),日间QC样本特征峰面积的RSD百分比低于30%的也达到了90.18%(图3c),说明该方法稳定性良好。
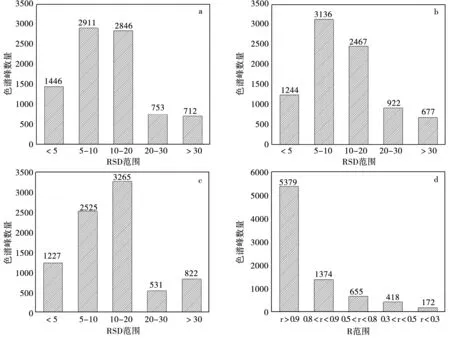
图3 UHPLC-Q-TOF MS/MS方法的评价Fig.3 Evaluation of UHPLC-Q-TOF MS/MS method
在非靶向UHPLC-Q-TOF MS/MS分析中,可能会存在一些结构未定的物质,所以很难采用常规方法对各物质进行线性范围的考察。本研究中称取20~160 mg样品,经50%甲醇提取分析后,计算各质谱特征峰的峰面积与样品量间的线性相关系数。结果发现(图3d)约有67.25%的代谢物响应与样品量间的线性相关系数(r)大于0.9,线性相关系数大于0.8的物质约占84%,说明在20~160 mg样品量范围内,大部分物质响应值与其对应样品量间线性关系良好,该方法比较稳定可靠。
2.3 方法的应用
为了检验已建立的UHPLC-Q-TOF MS/MS方法对大米代谢物的提取和分离效果,我们选取两种不同大米样品进行测定,检测两种稻米所得总离子流色谱图如图4所示。
通过比较仪器色谱图可以发现,前文中所建立的UHPLC-Q-TOF MS/MS方法对大米代谢物的提取效果和分离程度较好。稻米代谢物的组分极性范围较宽,从高极性到低极性的化合物均能在基峰色谱图中有所体现:在0~8 min主要集中的是极性大的组分;在12~18 min主要集中的是极性较小的部分。8~10min期间组分峰较少,可能是由于该时间段内化合物较少或组分离子化程度不高。UHPLC-Q-TOF MS/MS检测结果经色谱峰的提取,峰对齐,去噪音等处理,共得到9 146个特征峰,其中第一个稻米样品8 233个,第二个稻米样品8 756个,两种样品共有特征峰7 843个,占总特征峰的85.8%。后期可以通过质谱检索数据库检索结果进行匹配,并结合保留时间、特征离子、相对丰度以及相关文献对化合物进行鉴定和进一步的统计分析。
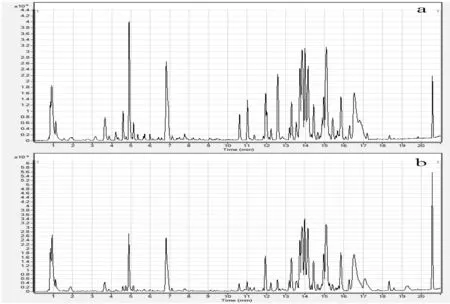
图4 两种稻米样本的总离子流色谱图Fig.4 Total ion chromatogram of rice samples
3 结论
本文通过对稻米代谢物的提取方法(包括提取试剂、提取料液比、提取方法及提取时间)和液相色谱分离方法(包括流动相洗脱和色谱柱温)进行筛选和优化,建立了利用UHPLC-Q-TOF MS/MS技术对稻米代谢物的分析方法。最终确定代谢物的最佳提取方式为;在料液比1∶5的情况下,50%甲醇经超声提取30 min可获得较好的提取效果。代谢物的最佳液相洗脱条件为30℃色谱柱温的条件下在乙腈-水-甲酸流动相体系以0.3 mL/min的流速梯度洗脱23分钟。该方法具有良好的重现性、日内/日间精密度以及线性关系。在此基础上,利用该方法对两种稻米样品进行分析,实验结果显示,共得到9 146个特征峰,其中第一个稻米样品8 233个,第二个稻米样品8 756个,两种样品共有特征峰7 843个,占总特征峰的85.8%,说明该方法对于大米代谢物的提取和分离稳定有效。在今后的研究中,可以通过色谱峰的比对和质谱检索数据库检索结果对大米代谢物进行识别和鉴定,进一步对液质联用所得结果进行分析。
