方镁石高压结构预测和高温结构稳定性研究*
2019-06-29宋婷孙小伟魏小平欧阳玉花张春林郭鹏赵炜
宋婷 孙小伟 魏小平 欧阳玉花 张春林 郭鹏 赵炜
(兰州交通大学数理学院,兰州 730070)
1 引 言
方镁石是地球下地幔矿物—镁方铁矿(Mg,Fe)O的主要组分,化学组成为氧化镁(MgO).在下地幔中,方镁石的含量仅次于钙钛矿(MgSiO3)[1],同时方镁石也是耐火陶瓷工业的基本材料,对其高温高压性质的研究具有重要的地球物理意义和重要的工业应用价值.由于MgO在凝聚态物理和地球科学研究中的重要性,其高温高压热力学性质已经通过静高压、动高压实验及计算机模拟方法进行了广泛的研究[2-5].尽管MgO材料的基础物理性质目前已经得到了较好的了解,但理论预测的熔化线和高压下实验测量结果之间存在巨大的分歧[3,6],为澄清歧见人们展开了对MgO高压结构的进一步研究[7,8];MgO高压相变研究以及基于各种可能新相结构的热力学特性的可靠性预测正逐渐成为材料高压科学和凝聚态物理领域的一个新课题.
有关MgO晶体的高压物性研究,早期主要集中在相变部分.MgO具有简单的立方岩盐结构(即NaCl结构,用B1表示),高压下会发生从岩盐结构到立方氯化铯结构(即CsCl结构,用B2表示)的相转变.1995年,Duffy等[9]的实验研究发现压力高达227 GPa时,岩盐结构的MgO依然稳定存在,和其后诸多理论计算的预测结果相一致[10-12].促使人们对MgO高压结构进行进一步深入研究的一个重要原因,是其理论预测和实验测量的熔化曲线之间存在很大的分歧,而不同理论方法的差别和实验系统误差不足以说明得到的熔化数据存在巨大分歧的原因.1994年Zerr和Boehler[13]给出了MgO压力仅上升到31.5 GPa的静高压实验熔化温度数据,测量得到的熔化线斜率为36 K/GPa,而2008年Zhang和Fei[3]的冲击波实验测量值为221 K/GPa,两者之间相差6倍多.分析误差产生的原因,除样品遭受了非静水压或热压力的影响外,还有可能是静高压装置中判断样品发生熔化的依据有问题,即对样品表面观测到的对流也许仅仅意味着样品表面而非样品内部发生了熔化;在冲击压实验中,Anderso和Duba[14]也提出过同样的问题,他们指出由Hugoniot声速测量结果计算的熔化温度中存在“过热”熔化现象,即晶体在高于自身熔点的温度下发生熔化的现象,但并没有对这个问题做进一步的定量分析;本课题组曾利用单相分子动力学模拟方法将0.1 MPa,31.5 GPa,47 GPa和135 GPa压力下的分子动力学热不稳定性数据和实验数据比较得到的过热率经过检验,用到了高压情况,外推得到了MgO达150 GPa的高压熔化曲线[15],结果远高于Zerr和Boehler[13]静高压实验给出的数据.
模拟地球深部的高温高压条件的实验手段主要有静高压和动高压实验技术.静高压技术由于受到金刚石压砧所能实现的压强上限的限制,测量能达到的压力范围有限,目前金刚石单晶压砧技术可以达到的最高压强约为350 GPa[16],但实验上100 GPa以上高压的实现和控制非常困难;采用冲击压缩技术虽然可以在比较高的压力和温度范围内研究材料的高压物性,但是由于冲击波传播速度很快,在样品中持续的时间很短,会给实际测量带来很多困难.Zhang和Fei[3]研究报道了单晶MgO在114和192 GPa冲击压缩下的雨贡纽数据,结合前人的冲击波数据分析了沿MgO的P-V雨贡纽线在(170 ± 10)GPa存在体积不连续的原因,认为是MgO发生从NaCl立方体结构到NiAs六角密堆积结构(用B81表示)的相变所引起的;Aguado和Madden[6]的模拟计算显示MgO从纤锌矿相(用B4表示)发生熔化的温度比从岩盐相发生熔化的温度低很多,可以解释高压下理论计算的熔化线和实验结果相差很大的矛盾.最近,尽管针对MgO的B81和B4结构,国际上开展了一些新的预测性研究工作[7,8],但MgO存在B81和B4新结构的合理性还需要进一步的论证.
众所周知,材料不同的结构必然导致不同的性质,MgO晶体各种热力学特性的可靠预测研究,其高压结构的细致研究是前提.本文利用基于密度泛函理论(DFT)的第一性原理计算方法,对MgO在0—800 GPa压力范围内的各种可能结构及利用基于粒子群优化算法的卡利普索(CALYPSO)晶体结构预测方法得到的潜在结构进行了系统研究,发现MgO在0—580 GPa的压力范围内一直以稳定岩盐结构存在,随着压力的增加,会发生从B1到B2结构的相转变;尽管B81和B4结构能合理解释冲击压缩实验中沿MgO的P-V雨贡纽线在(170 ± 10)GPa存在体积不连续和高压下理论计算的熔化线与实验结果相差很大的原因,但在压力高达800 GPa下都不能够稳定存在.在MgO高压结构预测基础上,本文分别利用壳层和呼吸壳层模型分子动力学方法,结合检验的有效经验势参数,在500—10000 K的温度范围和0—150 GPa的压力范围内对其稳定岩盐结构的熔化特性进行了模拟.
2 计算方法
2.1 MgO高压结构预测及优化
本文使用由吉林大学马琰铭小组开发的拥有自主知识产权的基于粒子群优化算法的CALYPSO软件包[17],预测MgO的1—4倍分子式组成的晶胞分别在0,135,300和500 GPa压力下的晶体结构.该软件只需给定化学配比,就能在特定的压力和温度条件下预测材料的基态和亚稳态结构.这种预测结构的方法已经成功应用在大量实验合成的单质和化合物的常压及高压晶体结构研究中[18].例如,2014年,Li等[19]利用该方法对H2S在10—200 GPa压力区间的结构进行预测,提出了两个能量稳定且具有金属性的新高压相P-1和Cmca,并首次预言高压下H2S的高温超导电性;在这一预测工作的启发下,Drozdov等[20]开展了S-H体系的高压超导实验研究,发现S-H体系在高压下呈现两个超导态,其中低温高压下测得的Tc与Li等[19]的计算数据基本吻合,而室温退火后测得的Tc达到惊人的203 K;2017年,Peng等[21]利用该方法预言了更多富氢包合物结构高压高温超导体的存在.本文对预测得到的结构采用第一性原理计算的VASP软件包进行几何优化[22],电子和电子之间的交换关联势采用广义梯度近似(GGA)下的Perdew-Burke-Ernzerhof泛函进行处理[23],计算过程中Mg的2p6,3s2和O的2s2,2p4电子均被看作价电子处理,价电子和芯电子之间的相互作用采用投影缀加平面波描述[24],倒易空间布里渊区k点采用Monkhorst-Pack方法选取[25],精确几何优化中的最大分割间隔为2π×0.08 Å—1.
获得MgO预测结构后,应用CASTEP软件包对预测结构、实验结构及其他可能存在的结构进行结构优化并计算相关性质[26],这里电子与电子间的交换关联能采用2008年由Perdew等[27]修订的Perdew-Burke-Ernzerhof形式的GGA方法,该形式的交换相关泛函能够提高密堆固体的平衡特性;芯电子与价电子间的相互作用势由超软赝势[28]实现.为了确定计算收敛性,文中对平面波截断能和k点采样进行了收敛性测试.针对MgO的B1,B2,B4等7种不同结构,截断能均取600 eV,就能保证高压下的收敛精度.零压和不同外压条件下的MgO晶体结构和原子位置优化采用Broyden,Fletcher,Goldfarb和Shanno提出并发展起来的几何算法[29].声子谱的计算采用线性响应方法[30].
2.2 分子动力学模拟技术
论文针对MgO岩盐结构,研究了高压下温度对该结构稳定性的影响.岩盐结构的MgO模拟体系由5×5×5个单位原胞组成,通过对多个单位原胞组成的超胞施加三维周期性边界条件,从而使离子的运动空间成为赝无限.粒子的初始速度根据模拟体系设定的温度赋值,压力由维里定理得到,模拟中长程静电力的处理采用Ewald求和技术[31],分别在实空间和倒空间中进行计算.模拟选用等温等压系综,即NPT系综.模拟时间步长设为1 fs,每个平衡态计算20000步(20 ps),前10000步(10 ps)是趋向平衡阶段,以使体系平衡到所设定的温度和压力,然后再计算10000步(10 ps),以测量指定压力下MgO模拟体系的温度和体积.
对MgO晶体进行经典分子动力学模拟研究,离子间相互作用势函数和可靠作用势参数的选取最为关键.本文中,MgO晶体各离子间的总相互作用能Vij由长程库仑能和短程非库仑作用能组成,短程相互作用取Buckingham形式:

(1)式右边各项分别表示库仑能、排斥能项和范德瓦耳斯项(偶极-偶极项),其中Z为有效电荷;r为原子之间的相互作用距离;A和ρ是排斥势参数;C为范德瓦耳斯常数.为了很好地描述离子的极化特性,模拟中考虑了广为使用的壳层模型[32],即认为每一个点离子由核和壳两部分组成,若核所带电荷为X,壳为Y,则这个点离子的总电荷为X+Y.相同离子的核和壳之间不存在静电相互作用,认为它们之间由一弹簧相连,这个弹簧的弹性常数为K;该离子在周围其他离子的电场中得以极化,设极化率为η,则η与该离子的壳电荷Y及弹性常数K有如下的关系式:

引入壳层模型能很好地描述离子的极化特性,模拟选用了两套非常相似的壳层模型势参数:Lewis和Catlow[33]给出的势参数(SM-LC)及Stoneham和Sangster[34]给出的势参数(SM-SS),来模拟MgO体系的熔化特性.
在壳层模型基础上考虑压缩效应就是呼吸壳模型,该模型允许氧离子的排斥半径在晶体中其他离子的作用下各向同性地变形,每个离子的核和呼吸壳层通过谐波势连接,因此,系统的总势能包括呼吸能量项Ubre:
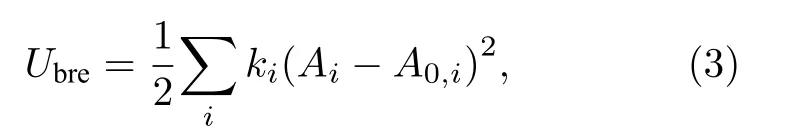
式中,ki是呼吸离子i的弹性常数;A0,i是自由离子i的排斥半径;Ai是呼吸壳层模型中允许离子i的排斥半径在晶体中其他离子作用下各向同性变形的排斥半径[35].这些短程对势参数、壳层电荷Y、描述壳层模型极化特性的弹性常数K和呼吸壳层模型中描述呼吸离子的弹性常数k通过拟合MgO晶体的结构参数得到.最近,Ball和Grimes拟合了一套新的MgO经验势参数(记为BG)[36],其中Mg和O离子仅带部分电荷(± 1.7e);正如Henkelman等[36]指出的:除非离子之间的距离非常短,否则Ball和Grimes拟合的MgO经验势参数[36]与Lewis和Catlow拟合的势参数[33]相比,差别很小.这些拟合势参数一并由表1和表2列出[33-36],以做全面比较.

表1 岩盐结构的MgO晶体特性模拟的短程对势参数Table 1.Parameters of the short-range pair potentials for simulation of MgO crystal properties with NaCl-type structure.

表2 岩盐结构的MgO晶体特性模拟的壳层及呼吸壳层参数Table 2.Shell and breathing shell parameters of MgO crystal characteristic simulation with NaCltype structure.
2.3 计算方法可靠性检验

图1 模拟得到的零温下MgO岩盐结构的体积比率随压力的变化及和实验结果的比较Fig.1.Volume ratios of MgO with NaCl-type structure under pressures at zero temperature,in comparison with the experimental data.
为了检验计算方法及选取参数的可靠性,本文利用壳层和呼吸壳层模型分子动力学方法及基于DFT的GGA及局域密度近似(LDA)方法,分别计算了零温下MgO岩盐结构从零到地球核幔边界压力(135 GPa)范围内的体积比率随压力的变化,所得结果与静压和非静压X射线衍射实验结果[2]及金刚石压砧(DAC)静高压装置得到的结果[37]进行了比较,如图1所示.在准静水压条件下,Fei[2]用氖做传压介质和氯化钠做压标测得了温度为300 K、压强上升到23 GPa时MgO的DAC静态压缩实验值,呼吸壳层模型分子动力学模拟的压力随体积比率变化的关系和Fei[2]上升到23 GPa时的静态数据吻合得很好,结合SM-LC势参数的壳层模型分子动力学模拟结果和Duffy等[9]整理的上升到100 GPa时的冲击压缩数据一致.同时,呼吸壳层模型分子动力学模拟结果和第一性原理GGA计算结果在高压下符合得很好,由于GGA和LDA方法上的差别,使得基于LDA的第一性原理计算往往低估晶格常数从而低估模拟体积,但这种低估在低压下不明显.和结合SM-SS势参数的壳层模型分子动力学模拟结果相比,呼吸壳层模型在描述MgO高压物态方程性质时,压缩效应体现的尤为明显.
3 分析和讨论
3.1 高压结构预测
常压下MgO以B1结构存在,高压下会发生从B1到B2结构的相变[9-12].Zhang和Fei[3]研究报道了单晶MgO在114和192 GPa冲击压缩下的Hugnonit数据,结合前人的冲击波数据分析了沿MgO的P-VHugnonit线在(170 ± 10)GPa存在体积不连续的原因,认为是MgO发生从B1立方体结构到B81六角密堆积结构的相变所引起的[3];Aguado和Madden[6]的模拟计算显示MgO从B4纤锌矿结构发生熔化的温度比从B1结构发生熔化的温度低很多,可以解释高压下理论计算的熔化线和实验结果相差很大的矛盾.

图2 MgO晶体的可能结构(其中大球代表Mg原子,小球代表O原子)(a)B1;(b)B2;(c)B3;(d)B4;(e)B81;(f)B82;(g)P3m1Fig.2.Probable crystal structures of MgO (the large and small spheres represent magnesium and oxygen atoms,respectively):(a)B1;(b)B2;(c)B3;(d)B4;(e)B81;(f)B82;(g) P3m1.
另外,还考虑了MgO的闪锌矿结构(用B3表示),这可以通过与其所在的碱土金属氧化物进行对比来说明.MgO是排在BeO之后CaO之前的碱土金属氧化物.本课题组曾对CaO的高压物性进行过研究[38,39],选择CaO的原因是它的B1→B2相变压力已由实验确定为60 GPa左右[40],可作为检验模拟计算准确与否的标准,而且CaO常压下B1结构到高压下B2结构的相变并无疑议.BeO不同于其他的碱土金属氧化物,结晶时形成的是稳定纤锌矿结构,而其他的碱土金属氧化物结晶时形成的是立方岩盐结构;根据Phillips电离度理论(电离度 ≥ 0.785的二元化合物在结晶时形成的是B1结构,而电离度 < 0.785的二元化合物形成的是B4结构或B3结构)[41],B4结构的BeO在高压下会转变为B1结构,而近年的理论研究表明,B4结构会先转变为B3结构,然后再转变为B1结构,van Camp和van Doren[42]预测了BeO晶体B4 → B3 → B1的相变压力分别为74和137 GPa,计算中采用了软的非局域赝势.直到最近,Cai等[43]采用第一性原理赝势和GGA研究了这两个相变的势垒,认为B4 → B3这一相变不可能发生,仅B4 → B1这一相变可以在高压下发生;Luo等[44]的最新计算表明随着压力增加到100 GPa,BeO直接由B4结构转变为B1结构.事实上,由于计算误差以及B3和B4结构的能量差十分接近(在零温零压时的能量差都在平均每个原子10 meV左右),简单的能量计算结果不能作为判断B3结构是否存在的有力证据.同时两种结构非常相似(B4结构是六角密堆形式,B3结构是立方密堆形式),导致这两种结构的物性也非常相似,其中之一就是非常接近的内能,这就解释了为什么在不同的计算中有时存在B3结构,有时不存在B3结构.最近Aguado和Madden[6]的模拟计算显示MgO从B4相发生熔化的温度比从B1相发生熔化的温度低很多,用来解释高压下熔化线的理论计算和实验测量之间存在巨大差别的原因.如果MgO和BeO一样,存在稳定的B4相,也应该存在一个和B4相非常相似的B3相.以上分析的MgO各种可能存在的结构B1,B2,B3,B4,B81及基于粒子群优化算法预测的晶体结构B82和P3m1的结构示意图由图2给出.
赝势平面波方法的一大优点就是它能够自动根据原子的受力情况来调整原子的位置和晶胞参数,直到所有原子的受力都为零,从而使整个体系的总能达到最低,找到给定条件下的最稳定结构.图3给出了利用平面波赝势结合GGA,DFT方法得到的MgO的7种可能结构的每个分子式的焓差随压力的变化,其中以B1结构作为参考.由于实验一般都是在一定温度T、压力P下进行的(由于热胀冷缩,使体积V固定的实验很难进行),所以严格来说此时应以吉布斯函数G判定相的稳定性.当温度为零时,G在数值上等于焓H,这时可通过热力学函数焓来判断热力学相的稳定性.本工作中所加外压均为流体静压力.在给定外压下,各相的相对稳定性可由焓H=E+PV来确定,焓最小的结构最稳定.可以看出零温下MgO最稳定的结构为B1结构,随着压力增加到580 GPa,MgO会发生从B1到B2结构的相转变,这符合压致相变中压力下的配位原则,即高压相的配位数(B2结构的配位数为8)大于等于低压相的配位数(B1结构的配位数为6);B2相是MgO的高压相,属体心立方结构,空间群为无实验晶格参数,模拟得到的晶格参数为2.660 Å;其他各相在0—800 GPa的压力范围内都不能够稳定存在.

图3 计算得到的MgO晶体在零温下的B1,B2,B3,B4,B81,B82和P3m1各可能结构的相对焓随压力的变化Fig.3.Calculated relative enthalpies of MgO with B1,B2,B3,B4,B81,B82andP3m1 phases as a function of pressure at zero temperature.
声子谱,即格波的角频率与波矢量的关系曲线,通过对晶体材料声子谱研究可以明确材料是否具有动力学稳定性特征;同时,根据声子谱中谱线重叠以及各原子声子态密度峰的位置可反映出原子间是否存在相似或相同的振动状态,从而能够推断出原子间是否存在相互作用以及作用力的强弱.为了检验本文提出的MgO晶体7种可能结构的动力学稳定性,在DFT框架下,采用线性响应方法[30]补充计算了零压下B1,B2,B3,B4,B81,B82和P3m1结构的声子谱和B2结构在相变压力580 GPa下的声子谱,如图4所示.可以看出,零压下MgO晶体B2结构的声子谱有虚频存在,说明该结构在这种状态下是不稳定的,当压力达到580 GPa时,B2结构成为稳定结构,符合前面热力学稳定性计算结果;零压下B3,B4,B81,B82和P3m1结构的声子谱在整个布里渊区均没有虚频出现,这意味着它们在零压下是动力学稳定的,是MgO的亚稳结构.
3.2 高温结构稳定性
固体发生熔化时,固、液两相的吉布斯自由能相等,而体积发生突变,且伴随着熵增,因此,属于一级相变.在熔化过程中,其抗剪切能力消失,在结构上由长程有序结构变为无序结构,固、液两相之间在晶体学上没有任何明确的位向关系,因此属于重构性相变.物质在极端压力条件下的熔化行为是一种非常复杂的物理过程,在宽广的温度和压强范围内,要对物质熔化等热力学性质做出合理描述,将涉及复杂的离子间的相互作用问题,选择考虑极化效应的壳层模型和在考虑极化效应基础上考虑压缩效应的呼吸壳层模型,模拟了零压下岩盐结构的MgO在500—5000 K温度范围内的体系的摩尔体积和总能随温度的变化,如图5和图6所示.
由图5和图6可以看出,岩盐结构的MgO在加热到一定温度时,对应的摩尔体积和总能发生了明显的跃变:利用呼吸壳层模型模拟得到的跃变温度为4490 K,利用壳层模型分子动力学且结合SM-SS和SM-LC势参数模拟得到的跃变温度分别为4495和3894 K,利用BG作用势参数模拟得到的跃变温度为3796 K,其中利用SM-LC和BG势参数模拟得到的结果非常接近.熔化是一个动力学过程,根据现代熔化理论,晶体的熔化温度可根据一定的熔化机制来进行修正[46],即过热率可按下式来修正:


图4 计算得到的MgO晶体B1 (a),B3 (b),B4 (c),B81(d),B82(e),P3m1 (f)和B2 (g)结构在零压下的声子谱和B2结构在相变压力为580 GPa下的声子谱(h)Fig.4.Calculated phonon spectra of MgO with B1 (a),B3 (b),B4 (c),B81(d),B82(e),P3m1 (f)and B2 (g)phases at 0 GPa and with B2 phase at 580 GPa (h).
式中,T为模拟观测到的温度;Tm为材料实际熔化温度.我们曾利用单相分子动力学模拟方法将0.1 MPa,31.5 GPa,47 GPa和135 GPa压力下的分子动力学热不稳定性温度和实验数据比较来确定过热率,得到的不同压力下的过热率基本一致[15].事实上,过热熔化跟升温速率有很大的关系,而压力对其影响不大,可据此来修正晶体过热熔化温度,本文模拟的加温间隔为100 K.和实验熔化温度3083 K进行比较[47],得到呼吸壳层模型模拟的过热率为0.456,壳层模型分子动力学结合SMSS和SM-LC势参数模拟得到的过热率分别为0.458和0.263,BG作用势参数模拟的过热率为0.231.根据晶体熔化均匀形核理论[46]和动力学灾变理论[48]定义的过热极限来看,过热发生在一个很大的范围之内,即:θ在0.1—2.0范围,本文得到的过热率符合该过热极限.从这个意义上来讲,本文选取的壳层和呼吸壳层模型均可用来研究MgO的高压熔化特性和高温结构稳定性.

图5 分子动力学模拟得到的零压下的MgO岩盐结构的体积随温度的变化Fig.5.Molecular dynamics calculated volume of MgO with NaCl-type structure as a function of temperature at zero pressure.
图7给出了单相分子动力学模拟得到的岩盐结构的MgO压力达150 GPa的熔化相图,该相图对模拟得到的热不稳定性温度结合过热率进行了修正,同时和Zerr和Boehler[13]利用DAC技术得到的实验数据以及Belonoshko和Dubrovinsky[49]的两相分子动力学模拟、Wang经验模型[50]与Lindemann熔化方程[51]得到的结果做了比较.为了消除“加热模拟体系直至熔化”方法产生的过热,Luo等[52]提出了“过热-过冷循环”方法,单相方法模拟固体熔化会引起过热,如果用单相方法来模拟液体结晶同样会导致过冷,过热与过冷的程度相当,因此如果采用单相模拟的方法,那么过热与过冷从体积变化图上将形成一个封闭的环,对过热与过冷进行适当的“平均”就可以定出熔化温度.“过热-过冷循环”方法即补充逆向液体结晶过冷模拟以消除过热的方法,实际上是对过热与过冷的一种平均,但其本质上也是单相模拟.两相模拟的主要思想是先构建一个固-液共存的系统,固体和液体之间存在交界面,然后固定压力把系统的温度升到接近熔化温度,如果此时的温度低于实际的熔化温度,那么液体部分就要逐渐结晶,固-液交界面就要向液体一边移动直至最终完全结晶,如果此时的温度高于实际的熔化温度,那么固体部分就要逐渐熔化,固-液界面就要向固体一侧移动直至完全熔化.和两相模拟相比,单相模拟方法本质上是一种“加热模拟体系直至熔化”的模拟方法,但是这种方法由于加热比较快,所以容易引起过热现象,即晶体在高于自身熔点的温度下发生熔化的现象.这里针对呼吸壳层模型、SM-SS和SM-LC壳层模型、BG作用势参数模型计算的热不稳定性温度采用的修正过热率分别为0.456,0.458,0.263和0.231.由图7可以看出,本文呼吸壳层模型模拟结果与Belonoshko和Dubrovinsky[49]的两相分子动力学模拟结果符合,壳层模型和BG作用势参数模型模拟结果和Wang经验模型[50]预测熔化温度符合程度达到140 GPa.Zerr和Boehler[13]的DAC实验结果明显偏低于所有理论结果,这是由于表面效应的原因造成的,与大块晶体相比,细微颗粒的熔点要低得多,原因是细微颗粒具有大的比表面积和表面能,在温度很低的情况下就能在表面发生熔化,而在DAC实验中观察到的熔化温度刚好是发生表面熔化时的温度.

图6 分子动力学模拟得到的零压下的MgO岩盐结构的总能随温度的变化Fig.6.Molecular dynamics calculated total energy of MgO with NaCl-type structure as a function of temperature at zero pressure.

图7 分子动力学模拟得到的MgO岩盐结构压力达150 GPa的熔化相图Fig.7.Molecular dynamics calculated melting phase diagram of MgO with NaCl-type structure as a function of pressure up to 150 GPa.
4 结 论
本文利用基于DFT的第一性原理计算方法,对MgO各种可能存在的结构B1,B2,B3,B4,B81及基于粒子群优化算法预测的晶体结构B82和P3m1的稳定性在0—800 GPa的压力范围内进行了系统研究,发现MgO在0—580 GPa的压力范围内一直以稳定岩盐结构存在,随着压力增加到580 GPa,MgO会发生从B1到B2结构的相转变.尽管B81结构能合理解释冲击压缩实验中沿MgO的P-V雨贡纽线在(170 ± 10)GPa存在体积不连续的原因,B4结构亦能合理解释高压下MgO理论计算的熔化线和实验结果相差很大的原因,但声子谱计算表明MgO晶体推测的B81和B4结构及其他结构在零压下仅为MgO晶体的亚稳结构.在确定MgO固-固相变物理图景的基础上,采用经典分子动力学方法,分别引入能很好描述离子极化特性的壳层模型和离子压缩效应的呼吸壳层模型,对MgO岩盐结构的高温稳定性进行了模拟研究,发现利用呼吸壳层模型模拟得到的热不稳定性温度为4490 K,利用壳层模型分子动力学且结合Stoneham和Sangster[34]给出的SMSS势参数及Lewis和Catlow[33]给出的SM-LC势参数模拟得到的热不稳定性温度分别为4495和3894 K.和实验熔化温度相比,呼吸壳层模型模拟的过热率为0.456,壳层模型分子动力学结合SMSS和SM-LC势参数模拟得到的过热率分别为0.458和0.263,符合根据晶体熔化均匀形核理论和动力学灾变理论定义的0.1—2.0的过热极限.MgO晶体岩盐结构压力达150 GPa的熔化相图计算表明利用呼吸壳层模型、壳层模型模拟得到的结果和文献给出的两相分子动力学模拟结果符合,DAC实验结果明显偏低于理论结果的原因是由于表面效应造成,即在实验中观察到的熔化温度是发生表面熔化时的温度而不是整个块体材料的真正熔化温度.
