Autoimmune hepatitis and IgG4-related disease
2019-06-27KosukeMinagaTomohiroWatanabeHobyungChungMasatoshiKudo
Kosuke Minaga, Tomohiro Watanabe, Hobyung Chung, Masatoshi Kudo
Abstract IgG4-related disease (IgG4-RD) is a chronic-fibroinflammatory disorder affecting a wide range of organs. Elevation of serum IgG4 concentrations and abundant infiltration of IgG4-expressing plasma cells are key diagnostic features of this autoimmune disease. Although common organ involvement of IgG4-RD includes the salivary glands, pancreas, and bile duct, hepatic involvement is less well established. Recently, five studies identified a subtype of autoimmune hepatitis(AIH), called IgG4-associated AIH (IgG4-AIH). IgG4-AIH is diagnosed based on significant accumulation of IgG4-expressing plasmacytes in the liver in patients who met the diagnostic criteria for classical AIH. Although four of the five reports regarded IgG4-AIH based on hepatic accumulation of IgG4-positive cells alone, one report diagnosed IgG4-AIH based on both hepatic accumulation of IgG4-positive cells and elevated serum concentrations of IgG4. IgG4-AIH diagnosed based on the latter criteria may be a hepatic manifestation of IgG4-RD whereas IgG4-AIH diagnosed based on the former criteria may be a subtype of AIH. In this review article, we summarize and discuss clinicopathological features of IgG4-AIH.
Key words: Autoimmune hepatitis; IgG4; IgG4-related disease; IgG4-associated autoimmune hepatitis
INTRODUCTION
Emergence and establishment of IgG4-related disease (IgG4-RD) has dramatically changed our view of autoimmune disorders involving the pancreas and biliary tract[1,2]. Autoimmune pancreatitis (AIP) is classified into type 1 AIP and type 2 AIP,with the former type being regarded as a pancreatic manifestation of systemic IgG4-RD[3]. Moreover, a subtype of IgG4-RD preferentially affecting the biliary tract is now called as IgG4-related sclerosing cholangitis (IgG4-SC)[4]. IgG4-RD is a new disease entity characterized by elevated concentrations of serum IgG4 and infiltration of IgG4-expressing plasma cells in the affected organs. In addition to enhanced IgG4 antibody responses, many patients with IgG4-RD exhibit multiple organ involvement[5]. Clinicopathological analysis showed that IgG4-RD can occur in almost all the organs in the body, however, this disorder preferentially affects the pancreas,salivary glands, biliary tract, kidney, and lung[5].
Although type 1 AIP and IgG4-SC are recognized as pancreatic and biliary tract manifestations of systemic IgG4-RD[1-4], the hepatic involvement of this autoimmune disorder remains poorly understood[6]. Five studies including ours, have proposed a novel disease entity called IgG4-associated autoimmune hepatitis (IgG4-AIH)[7-11]. In this review article, we try to clarify clinical characteristics of IgG4-AIH by comparison with the characteristics of classical AIH. Furthermore, we discuss whether IgG4-AIH should be considered as a subtype of AIH or as hepatic involvement of systemic IgG4-RD.
HEPATIC INVOLVEMENT OF IGG4-RELATED DISEASE
Umemura et al[12]were the first to perform clinicopathological analysis of hepatic manifestations in IgG4-RD. They examined liver biopsy specimens obtained from 17 patients with AIP. Elevation of serum aspartate aminotransaminase (AST) and/or alanine aminotransaminase (ALT) was noted in 59% of AIP patients, whereas elevation of serum alkaline phosphatase (ALP) and/or gamma-glutamyl transferase(γGTP) was noted in 100%. Elevation of serum concentrations of IgG4, IgG, and antinuclear antibody (ANA) were seen in 94%, 65%, and 47%, respectively. Thus, AIP patients analyzed in this study shared biochemical and immunological abnormalities with AIH patients in terms of elevations of liver enzymes, IgG, and ANA[13-15]. A wide variety of histological findings, including portal inflammation (35%), interface hepatitis (24%), lobular hepatitis (29%), bile duct damage (59%), and canalicular cholestasis (53%) were observed in the livers of patients with AIP[12]. These pathological findings strongly indicate that around 20-30% patients with AIP exhibit pathological findings similar to classical AIH since portal inflammation, interface hepatitis, and lobular hepatitis are detected in most cases of AIH[13-15].
Infiltration of IgG4-expressing plasma cells in the liver was confirmed in 47% of patients with AIP, but not in those with classical AIH, primary biliary cholangitis(PBC), or primary sclerosing cholangitis (PSC) when more than 5 positive cells [/high powered field, (HPF)] was defined as positive[12]. The numbers of hepatic IgG4-expressing plasma cells were positively correlated to serum concentrations of IgG4 and the presence of bile duct thickness, suggesting that, in this study, patients with IgG4-RD involving both the pancreas (AIP) and the biliary tract (IgG4-SC) exhibit accumulation of IgG4-expressing plasma cells in the liver. These seminal studies by Umemura et al[12]provide evidence that a significant proportion of patients with AIP exhibit laboratory and pathological findings akin to classical AIH. At present, it remains unknown whether AIH-like liver lesions observed in patients with AIP are regarded as hepatic manifestation of IgG4-RD.
Another type of hepatic manifestation of IgG4-RD is hepatic inflammatory pseudotumor. Hepatic pseudotumors are classified into two types; fibrohistiocytic and lymphoplasmacytic[16]. Massive infiltration of IgG4-expressing plasma cells accompanied by obliterative phlebitis, one of the characteristic pathological findings in IgG4-RD[1,2], was seen in the lymphoplasmacytic type of hepatic pseudotumor.
Based on these pathological findings, Zen et al[16]proposed that the lymphoplasmacytic type of hepatic pseudotumor is a hepatic manifestation of IgG4-RD.However, detailed examinations of clinical characteristics, including serum IgG4 concentrations are lacking in this study in which 16 cases of hepatic pseudotumors were enrolled. Taken together, recent studies provide evidence that AIH-like lesions and hepatic pseudotumors might occur as hepatic involvement of systemic IgG4-RD.
IGG4-AIH AND CLASSICAL AUTOIMMUNE HEPATITIS
The first case of IgG4-AIH was reported by Umemura et al[17]in 2007. Serum levels of ALT, ALP, γGTP, IgG, and IgG4 were markedly elevated in this case. Interface hepatitis, lobular hepatitis, and rosette formation were seen in the liver biopsy specimens. This case met the diagnostic criteria for AIH proposed by the International Autoimmune Hepatitis Group (IAIHG)[18]and was diagnosed as AIH. On the other hand, this case was atypical with regard to classical AIH in that abundant infiltration of IgG4-expressing plasma cells in the portal tract and a marked elevation of serum IgG4 concentrations were observed. Based on enhanced IgG4 antibody responses, the authors proposed a new disease entity called IgG4-AIH[17]. Following this case report,five groups, including ours, tried to identify and characterize IgG4-AIH in patients who met the diagnostic criteria for AIH (Table 1)[7-11].
CLINICAL CHARACTERISTICS OF IGG4-AIH
We reviewed five reports addressing the incidence, laboratory and pathological findings, and efficacy of glucocorticoid treatment in IgG4-AIH[7-11](Table 1).
Diagnostic criteria and proportion of IgG4-AIH
Chung et al[7]were the first to examine the incidence of IgG4-AIH in patients with classical AIH. They examined 26 AIH patients who met the diagnostic criteria proposed by the IAIHG. They defined IgG4-AIH when more than 5 IgG4-expressing plasma cells (/HPF) were detected in the portal tracts. According to this criteria, 9 of the 26 AIH patients were diagnosed with IgG4-AIH[7]. In another study conducted in India, 10 of the 40 AIH patients were diagnosed as IgG4-AIH based on the Chung's criteria[7,9]. In subsequent three studies, IgG4-AIH was defined when more than 10 IgG4-expressing plasma cells were detected in the liver[8,10,11]. In one study, the presence of the overlap syndrome with IgG4-AIH in combination with PSC or PBC was reported[10]. Although the proportion of IgG4-AIH was highly variable (from 3.3% to 34.6%) probably due to the difference of the diagnostic criteria[7-11], it was clear that a unique type of AIH characterized by significant infiltration of IgG4-expressing plasma cells exists in patients with formerly diagnosed AIH. Moreover, Aydemir et al[11]reported that 6 out of 40 children with AIH were regarded as IgG4-AIH.Collectively, these five reports successfully identified a subtype of AIH, called IgG4-AIH, based on significant infiltration of IgG4-expressing plasma cells in the liver.
Nakanuma et al[19,20]proposed new diagnostic criteria for IgG4-AIH with reference to systemic IgG4-RD. According to the Nakanuma's criteria, IgG4-AIH is characterized by elevated concentrations of serum IgG4 and infiltration of IgG4-expressing plasma cells in the liver (> or = 10 cells /HPF). Although Umemura's study fully meets this strict criteria[8], the other four studies do not satisfy this criteria[7,9-11]. Application of this new criteria for the diagnosis might make IgG4-AIH an extremely rare disease entity since only three cases have met this criteria[19]. Given the fact the Nakanuma's criteria is proposed based on the well-established criteria for IgG4-RD, application of this criteria might be useful for the diagnosis of IgG4-AIH as hepatic involvement of systemic IgG4-RD. However, it remains to be determined whether IgG4-AIH occurs as a subtype of classical AIH or hepatic involvement of IgG4-RD.
Liver enzyme abnormality
Regarding liver enzyme abnormality, one study described that mean AST level in the IgG4-AIH group was significantly higher than in the classical AIH group whereas the differences of mean ALT, ALP and total bilirubin levels were not significant between the two groups[9]. In the remaining four reports[7,8,10,11], serum levels of hepatobiliary enzymes such as AST, ALT, ALP, and γGTP were comparable between IgG4-AIH and classical AIH (IgG4-non-associated AIH). Thus, blood biochemical examinations do not distinguish IgG4-AIH and IgG4-non-associated AIH in most cases.
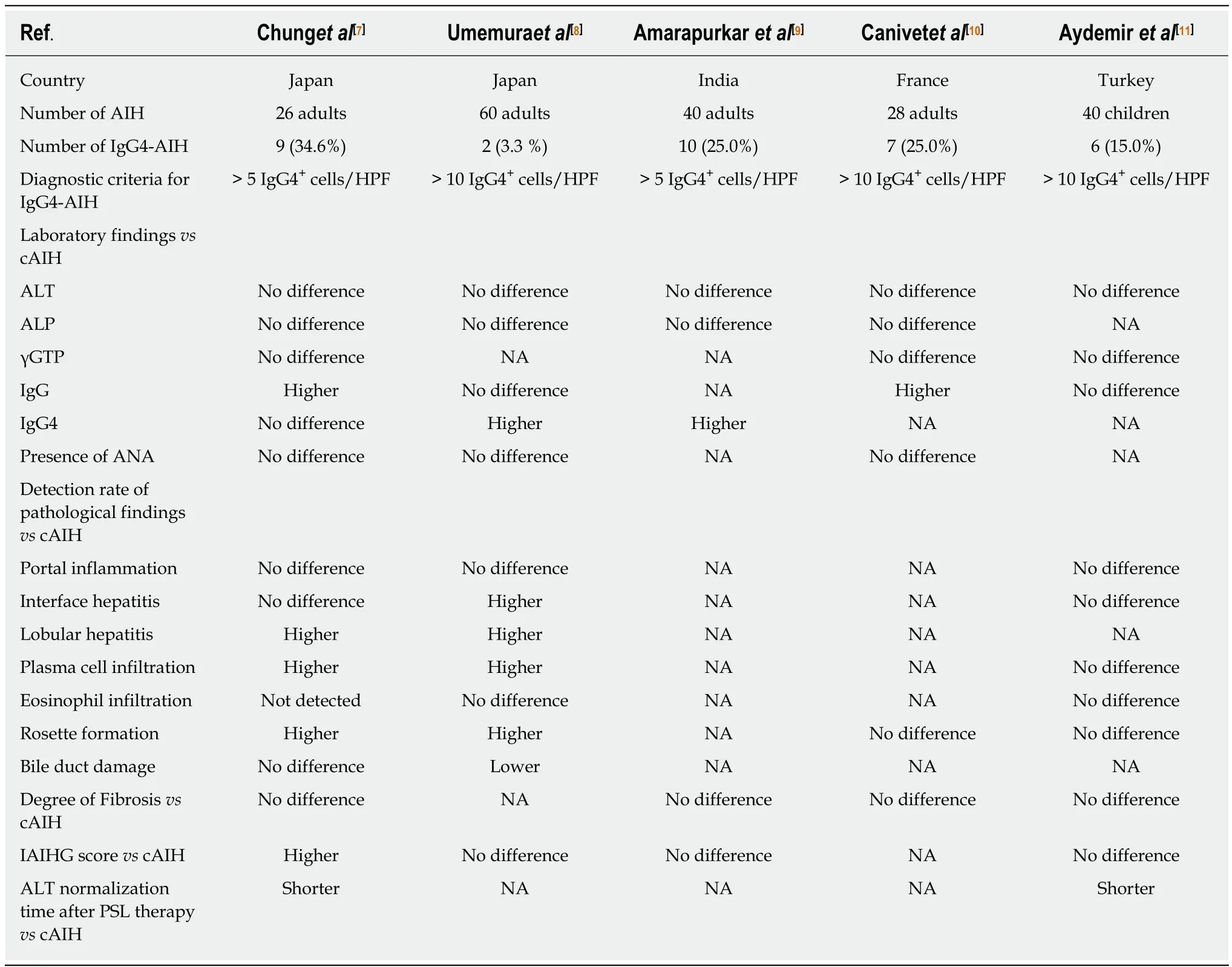
Table 1 Autoimmune hepatitis and lgG4-associated autoimmune hepatitis
Serum immunological findings
HLA DR-3 and DR-4 serotypes are associated with classical AIH[13-15]. Umemura et al.showed that one of two patients with IgG4-AIH was positive for HLA-DR4[8]. The HLA status was not examined in the other four studies[7,9-11]. Thus, it remains unknown whether HLA typing is useful to differentiate between IgG4-AIH and classical AIH.
Elevated levels of serum IgG and ANA titers are hallmarks of AIH[13-15]. No difference was seen in serum levels of ANA titers in these two types of AIH[7,8,10]. In contrast, serum concentrations of IgG were significantly higher in patients with IgG4-associated AIH than in those with IgG4-non-associated AIH in two studies[7,10].
Serum concentrations of IgG4 were measured in three studies[7-9]. Serum concentrations of IgG4 were comparable between the two types of AIH in one study[7]whereas patients with IgG4-AIH exhibited a marked elevation of this Ig subtype as compared with those with IgG4-non-associated AIH in the other two studies[8,9]. At present, the reasons accounting for the discrepancy between Chung's and Umemura's studies remain unknown. One plausible explanation is that IgG4-AIH defined by Chung et al[7]might be a different disease to that identified by Umemura et al[8]Considering the fact that Umemura's cases of IgG4-AIH meet the strict diagnostic criteria[19], IgG4-AIH defined by Umemura et al[8]might be a hepatic manifestation of systemic IgG4-RD rather than a subtype of AIH. On the contrary, IgG4-AIH defined by Chung et al. might be a subtype of AIH characterized by moderately increased IgG4 responses in the liver, but not in the serum.
Pathological findings
In Umemura's study[8], both of the patients with IgG4-AIH exhibited portal inflammation, interface hepatitis, lobular hepatitis, plasma cell infiltration, and rosette formation whereas neither presented with bile duct damage. Consistent with this report, the incidence of lobular hepatitis, plasma cell infiltration, and rosette formation was higher in patients with IgG4-AIH than in those with IgG4-nonassociated AIH[7]. Moreover, the degree of portal inflammation was more severe in patients with IgG4-AIH than in those with IgG4-non-associated AIH[7]. Thus, adult patients with IgG4-AIH appear to exhibit a more severe pathology than those with IgG4-non-associated AIH although the degree of fibrosis was comparable between the two disorders. In contrast to these studies in adult AIH patients, the degree of chronic inflammation in children did not show a significant difference between IgG4-associated AIH and IgG4-non-associated AIH as judged by the grade of portal inflammation, interface hepatitis, rosette formation, and fibrosis[11].
As for the type of immune cells characterizing IgG4-AIH, a marked infiltration of CD3+T cells, CD20+B cells, IgG+cells and CD38+plasma cells was seen in the liver of patients with IgG4-AIH as compared with that of patients with IgG4-non-associated AIH[7]. Abundant infiltration of these adaptive immune cells, especially plasma cells,leads us to speculate that enhanced IgG4 antibody responses can be partially explained by an increased accumulation of plasma cells in the liver and that moderately increased IgG4 antibody responses (5-10 cells/HPF) may be an epiphenomenon associated with increased accumulation of plasma cells[7,19]. However,infiltration of IgG1-expressing plasma cells was comparable in the liver between IgG4-AIH and IgG4-non-associated AIH in Chung's study[7]. Therefore, selective enhancement of IgG4 antibody responses might be also induced even in cases exhibiting normal serum IgG4 concentrations and moderately increased accumulation of IgG4-expressing plasma cells in the liver. In this regard, there is no doubt that selective enhancement in IgG4 antibody responses are actually induced in IgG4-AIH patients who met the strict criteria proposed by Nakanuma et al[8,19].
Sensitivity to glucocorticoid and azathioprine
Glucocorticoid treatment is the first line of treatment for both AIH[13-15]and IgG4-RD[1-4]. Sensitivity to glucocorticoids was investigated in IgG4-AIH and IgG4-nonassociated AIH through assessment of ALT normalization time. The proportions of patients showing normalization of ALT were comparable between IgG4-AIH and IgG4-non-associated AIH at 1 or 2 years after the initiation of glucocorticoid treatment[9,10]. In contrast, the ALT normalization time after the glucocorticoid treatment was shorter in patients with IgG4-AIH than in those with IgG4-nonassociated AIH in two studies[7,11]. Thus, accumulation of IgG4-expressing plasma cells in the liver might be a surrogate marker for the prediction of glucocorticoid treatment.Patients with IgG4-AIH were treated with glucocorticoid followed by the maintenance therapy with azathioprine[9-11]. The response rates to azathioprine at the maintenance phase were comparable between IgG4-AIH and IgG4-non-associated AIH.
IGG4-AIH AS A SUBTYPE OF AIH OR AS A HEPATIC MANIFESTATION OF IGG4-RD
One major question arising from the previous studies summarized above[7-11]is whether IgG4-AIH is a subtype of classical AIH or a hepatic manifestation of systemic IgG4-RD. Since all of these studies identified and characterized IgG4-AIH among patients with classical AIH, AIH is likely to be classified into IgG4-AIH and IgG4-nonassociated AIH. Given the fact that IgG4-AIH is diagnosed based on the presence of IgG4-expressing plasma cells in the liver, it is possible that immune responses necessary for IgG4 antibody class switch recombination underlie the immunopathogenesis of IgG4-AIH. Extensive immuno-histochemical analysis by Chung et al.showed that the numbers of CD3+T cells, CD20+B cells, and CD38+plasma cells in the liver were greater in patients with IgG4-AIH than in those with IgG4-non-associated AIH[7]. The results of this study strongly suggest that adaptive immune responses might be involved in the immuno-pathogenesis of IgG4-AIH and that enhanced IgG4 responses in the liver might be secondary and as a result of activation of adaptive immune cells. However, hepatic immune cell populations or profiles of cytokines leading to enhanced IgG4 antibody responses have been poorly clarified.
IgG4-RD is characterized by multiple organ involvement and a unique form of fibrosis called storiform fibrosis[1,2]. Although systemic IgG4-RD preferentially affects the salivary glands, pancreas, biliary tract, and kidneys[1,2], hepatic involvement is less well characterized[6]. Neither multiple organ involvement nor the presence of storiform fibrosis had been examined in patients with IgG4-AIH when these five studies were published[7-11]. Lack of multiple organ involvement as well as storiform fibrosis supports the idea that IgG4-AIH is a subtype of classical AIH rather than hepatic manifestation of IgG4-RD. However, it is too early to recognize IgG4-AIH as a subtype of classical AIH as a case with co-occurrence of IgG4-SC and IgG4-AIH has been reported[21]. Moreover, metachronous development of IgG4-SC and IgG4-related AIP was observed in two cases that had been formerly diagnosed as IgG4-AIH[19].These two cases of IgG4-AIH bearing other organ involvement and elevated serum concentrations of IgG4 are considered to exhibit AIH lesions as hepatic involvement of systemic IgG4-RD. Thus, IgG4-AIH can occur as hepatic involvement of systemic IgG4-RD if accumulation of IgG4-expressing plasma cells in the liver is accompanied by a marked elevation of serum IgG4 concentrations. Therefore, it is strongly suggested that IgG4-AIH identified in the previous five studies is categorized into two types[7-11]; a subtype of AIH characterized by moderately enhanced IgG4 responses in the liver alone[7]and a hepatic manifestation of IgG4-RD sharing pathological finding with AIH and exhibiting systemic enhanced IgG4 responses[8,19].
Another important question that needs to be addressed is the clinicopathological difference between IgG4-AIH and IgG4-hepatopathy, both of which can arise from systemic IgG4-RD[8,12,19]. Although IgG4-hepatopathy and IgG4-AIH share key pathological findings such as portal inflammation, interface hepatitis, and lobular hepatitis, the former disease frequently presents with bile duct damage and eosinophilic infiltration, compared to the latter[8,12,19]. Thus, careful pathological examinations might be useful in discriminating between these two disorders.
Much progress has been made in the immuno-pathogenesis of IgG4-RD in terms of autoantigens and pathogenic cell types. Laminin 511, Annexin A11, and galectin 3 are identified as target autoantigens in IgG4-RD[22-24]. Plasmacytoid dendritic cells (pDCs)producing both IFN-α and IL-33 play crucial roles in murine experimental AIP and human IgG4-related AIP[2,25,26]. Examination of serum antibody titers against the autoantigens listed above and analysis of pDC activation in the liver may be useful in determining whether IgG4-AIH occurs as a hepatic manifestation of IgG4-RD.
CONCLUSION
Recent clinicopathological analysis identified IgG4-AIH as a subtype of AIH.Confirmation of hepatic accumulation of IgG4-expressing plasma cells is absolutely required for the diagnosis of IgG4-AIH on the condition that the patient meets the diagnostic criteria for AIH. IgG4-AIH can occur not only in adults but also in children.Previous studies with a limited number of patients have indicated that IgG4-AIH and IgG4-non-associated AIH share laboratory findings, pathological findings, and sensitivity to glucocorticoids. However, accumulation of IgG4-expressing plasma cells was only detected in IgG4-AIH. It should be noted, however, that ALT normalization time after the glucocorticoid treatment might be shorter in IgG4-AIH than in IgG4-non-associated AIH[7,11]. Thus, significant infiltration of IgG4-expressing plasma cells in the liver may be useful as a biomarker for the prediction of the efficacy of glucocorticoid in AIH. One important issue that needs to be addressed in future studies is whether IgG4-AIH is a subtype of AIH or a hepatic manifestation of IgG4-RD. Establishment of IgG4-AIH as a new disease entity awaits characterization of clinicopathological findings and immune responses in accumulated cases with IgG4-AIH.
杂志排行

World Journal of Gastroenterology的其它文章
- Cyst fluid glucose: An alternative to carcinoembryonic antigen for pancreatic mucinous cysts
- Mechanisms of hepatocellular carcinoma progression
- Congenital peritoneal encapsulation: A review and novel classification system
- Electroacupuncture at ST36 modulates gastric motility via vagovagal and sympathetic reflexes in rats
- Role of D2 gastrectomy in gastric cancer with clinical para-aortic lymph node metastasis
- Risk factors for progression to acute-on-chronic liver failure during severe acute exacerbation of chronic hepatitis Β virus infection