Mechanisms of hepatocellular carcinoma progression
2019-06-27OlorunseunOgunwobiTrisheenaHarricharranJeannetteHuamanAnnaGaluzaOluwatoyinOdumuwagunYinTanGraceMaMinhhuyenNguyen
Olorunseun O Ogunwobi, Trisheena Harricharran, Jeannette Huaman, Anna Galuza,Oluwatoyin Odumuwagun, Yin Tan, Grace X Ma, Minhhuyen T Nguyen
Abstract Hepatocellular carcinoma (HCC) is the most common primary malignancy of the liver. It is the second leading cause of cancer-related deaths worldwide, with a very poor prognosis. In the United States, there has been only minimal improvement in the prognosis for HCC patients over the past 15 years. Details of the molecular mechanisms and other mechanisms of HCC progression remain unclear. Consequently, there is an urgent need for better understanding of these mechanisms. HCC is often diagnosed at advanced stages, and most patients will therefore need systemic therapy, with sorafenib being the most common at the present time. However, sorafenib therapy only minimally enhances patient survival. This review provides a summary of some of the known mechanisms that either cause HCC or contribute to its progression. Included in this review are the roles of viral hepatitis, non-viral hepatitis, chronic alcohol intake, genetic predisposition and congenital abnormalities, toxic exposures, and autoimmune diseases of the liver. Well-established molecular mechanisms of HCC progression such as epithelial-mesenchymal transition, tumor-stromal interactions and the tumor microenvironment, cancer stem cells, and senescence bypass are also discussed. Additionally, we discuss the roles of circulating tumor cells,immunomodulation, and neural regulation as potential new mechanisms of HCC progression. A better understanding of these mechanisms could have implications for the development of novel and more effective therapeutic and prognostic strategies, which are critically needed.
Key words: Hepatocellular carcinoma; Viral/non-viral hepatitis; Alcohol consumption;Epithelial-mesenchymal transition; Tumor-stromal interactions; Tumor microenvironment; Cancer stem cells; Circulating tumor cells; Immunomodulation;Neural regulation
INTRODUCTION
Hepatocellular carcinoma (HCC) is the most common primary liver cancer comprising 75%-85% of cases of liver cancer[1]. It is the sixth most common cancer and the second leading cause of cancer deaths worldwide[1]. The incidence of HCC in the United States has been increasing over the past two decades[1-3]. While the overall prognosis for HCC patients in the United States has improved somewhat in the past 15 years, it still remains poor. In fact, in the United States, the 2-year survival for HCC is less than 50% and 5-year survival is only 10%[4].
In Asia, chronic hepatitis B virus (HBV) infection is the primary cause of HCC.While in the Western world, chronic hepatitis C virus (HCV), alcoholic cirrhosis and non-alcoholic steatohepatitis (NASH) are the main causes[5]. Other known risk factors of HCC include heavy alcohol consumption, nonalcoholic fatty liver disease,consumption of aflatoxins, obesity, type 2 diabetes and tobacco smoking[6,7].
Early diagnosis and effective treatment of HCC remain a challenge. While some patients can be symptomatic, including symptoms such as right upper abdominal quadrant pain, anorexia, early satiety, weight loss, obstructive jaundice, fever, watery diarrhea, lethargy, and bone pain (from metastases)[6,7], most patients remain asymptomatic, and clinical presentation occurs at advanced stages of the disease.
If detected very early, HCC can actually be cured with an excellent long-term prognosis[7], where the principal treatment options would be surgical resection or liver transplantation if the patient is a suitable transplant candidate[8]. However, for the vast majority of HCC patients, their cancer is detected at an advanced stage where surgical cure is no longer an option[7]. Most patients will therefore need chemotherapy, which works by destroying cancer cells and inhibiting the proliferation of new cancer cells via the use of chemical agents. Sorafenib, a small multi-tyrosine kinase inhibitor that blocks Raf kinase, vascular endothelial growth factor (VEGF), and platelet-derived growth factor (PDGF) receptor activities, is the most commonly used chemotherapeutic agent to treat HCC[4]. Although a targeted chemotherapeutic agent, its use has been shown to minimally enhance patient survival[9]by only about 7-10 months[10]. Other drugs such as sunitinib, brivanib, and other angiogenic inhibitors are currently still under development and hold promise in targeting the extensive angiogenic network that is present in the liver[11,12]. Additional multi-kinase inhibitors recently approved for HCC treatment include regorafenib (for secondary treatment after sorafenib), as well as levatinib (another first-line drug to treat HCC besides sorafenib). However, neither provide much more additional benefit than sorafenib treatment[13,14]. As such, better treatment options are still needed.
To address this unmet need, researchers are trying to identify different mechanisms that may be involved in HCC progression to find alternative therapeutic strategies[8].There have been various signaling pathways and molecules implicated in HCC progression. Some of these will be discussed in this review article and are summarized in Figure 1.
MECHANISMS OF ETIOLOGY
Several risk factors have been implicated in the development and progression of HCC, notably chronic viral hepatitis, non-viral hepatitis, chronic alcohol intake,certain disease states (obesity and diabetes), and consumption of toxin-contaminated staples[15]. The epidemiologic distribution of these risk factors varies according to geographic location and host-specific factors.
Viral hepatitis
HBV and HCV are major causes of viral hepatitis that lead to the development of cirrhosis and HCC. The pathogenesis of HBV-induced HCC is thought to involve several mechanisms, including HBV-DNA integration into host genetic machinery,DNA methylation, oxidative stress, and HBx protein[16]. The risk of developing HCC has been shown to be proportional to HBV-DNA level in liver cells. HBV gains entry into liver cells through a receptor mediated pathway. Chronic illness results from persistence of the virus in the host cells via various mechanisms that include infection of immune defense control centers, viral inhibition of antigen presentation, selective immune suppression, down-regulation of viral gene expression, and viral mutations that functionally incapacitate virus-specific T cells from recognizing HBV antigen[17].Immune response and inflammatory reactions induce cytokine and chemokine mobilization, causing oxidative stress. This, in turn, promotes constant activation of several genes that cause cirrhosis, including TERT, MLL4, RARβ, CCNE1, Cyclin A2,FN1, ROCK1, SENP5, ANGPT1, PDGF receptor, calcium signaling-related genes,ribosomal protein genes, epidermal growth factor receptor (commonly known as EGFR), and mevalonate kinase carboxypeptidase[15].
HBV and HCV viral proteins may be involved in hijacking the cellular machinery.Viral attack can also directly cause cirrhotic tissue development through the release of proinflammatory cytokines (e.g., interleukin (IL)6, tumor necrosis factor (TNF)-α, IL1 and IL18)[18].
HCV hijacks host cellular machinery to increase cellular proliferation, steatosis,inflammatory processes, mitochondrial dysfunction, insulin resistance, all leading to oxidative stress, genetic instability and DNA damage with cirrhosis and HCC as a likely outcome[19].
HCC risk drastically increases at the cirrhotic liver stage, suggesting a close association.
The corresponding interplay of inflammatory responses, gene activation, and viral clearance suppression creates a conditioned environment that promotes cellular mutations leading to HCC.
Non-viral hepatitis
Even though viral hepatitis from HBV and HCV are strongly associated with liver cancer, there are non-viral risk factors that can induce the development of HCC[20].Diabetes mellitus, alcohol abuse, cardiovascular disease, liver inflammation, obesity,dyslipidemia and non-alcoholic fatty liver disease (NAFLD) are some other major contributors to HCC development.
Accumulation of iron in the liver of NASH and HCC patients[21,22]is correlated with progression of fibrosis and HCC[23]. In this context, a possible tumor biomarker may be serum ferritin rather than iron. However, because there is no exact correlation between iron inside the liver and iron in the blood, it is difficult to clarify the pathological features of ferritin on the poor prognosis of non-viral HCC (nvHCC)[24].Results of a cohort study of 93 patients with nvHCC, 62 of whom had alcohol abuse problems, showed an increase in ferritin level in non-diabetics[24]. However, further research needs to be done to assess the correlation between the impacts of alcohol and ferritin on NAFLD[24].
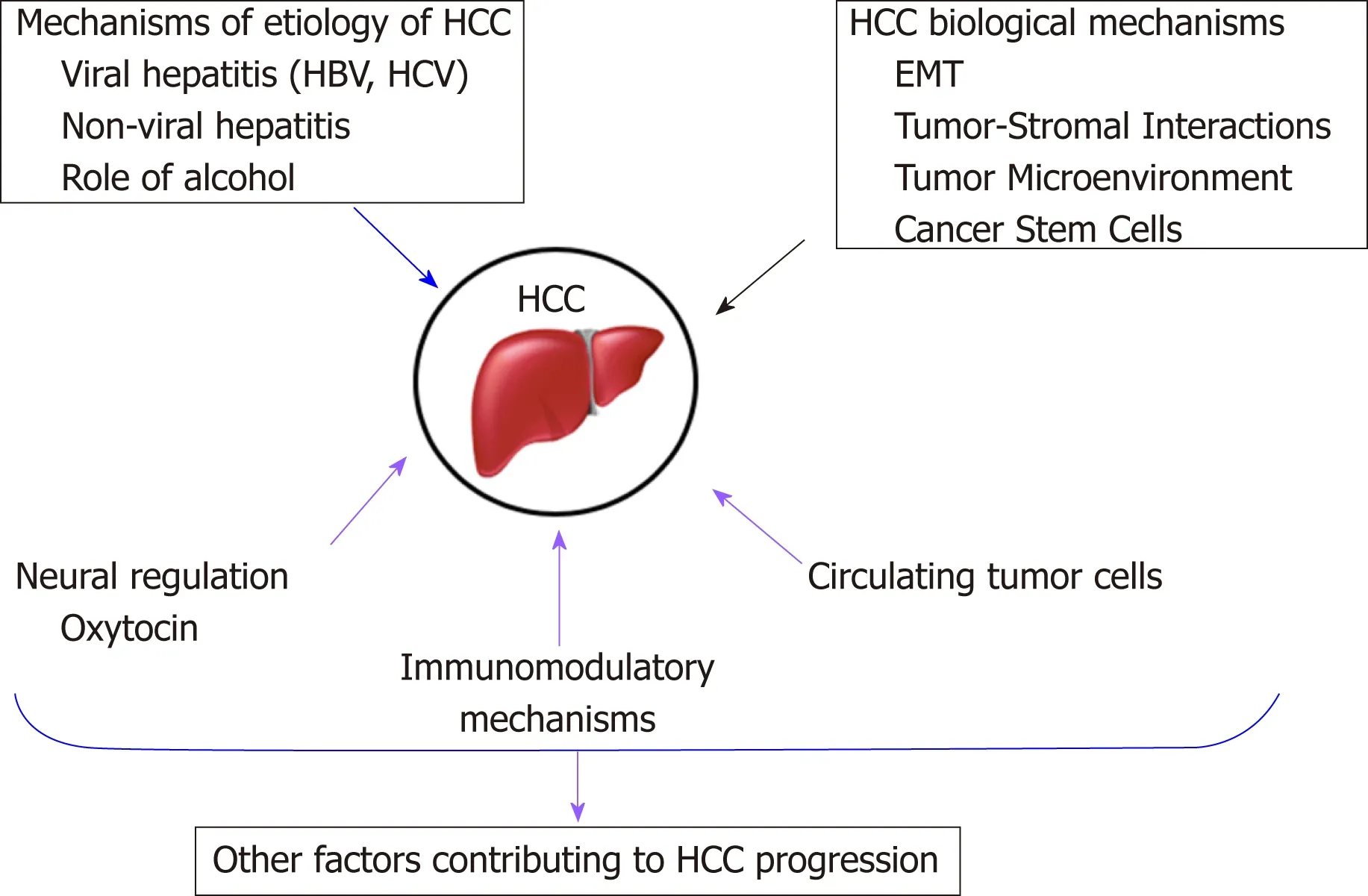
Figure 1 Summary of the HCC progression mechanisms discussed in this review.
On the other hand, HCC is associated with obesity. Obesity impairs metabolism,induces inflammation and is an etiological factor for NAFLD, steatosis, NASH,hepatic fibrosis, cirrhosis, and ultimately HCC. Caused partly by a sedentary lifestyle and obesity, impaired lipid metabolism and deregulation of energy equilibrium in the liver contributes to the correlation between type 2 diabetes and NAFLD. In fact,several studies have shown that high BMI, waist circumference, and type II diabetes mellitus are associated with higher risks of liver cancer[25,26]. They have also suggested that the association may vary depending on the status of viral hepatitis infection[25].Conversely, NAFLD provides the metabolic environment to induce insulin resistance[27], a known etiological factor for HCC.
Role of alcohol
Chronic alcohol intake is detrimental to our health. It leads to liver cirrhosis, and subsequently HCC. Alcoholic liver disease is one of the leading causes of HCC[28].According to case studies from all over the world, alcohol abuse is related to up to 2-fold increased risk of HCC[29]. Moreover, studies performed on mice fed an alcohol diet have shown exacerbation of inflammation, epithelial-mesenchymal transition(EMT) and fibrosis, and consequent progression to HCC[28].
Pure ethanol does not directly cause inflammation and liver damage, however,toxic by-products of alcohol catabolism such as accumulation of acetaldehyde and free radicals can influence oxidative stress, apoptotic cell death, necrosis and necroptosis[29]. Reactive oxygen species (ROS) generation is the result of increased inflammatory cytokine secretion caused by constant inflammatory pathways[19]. ROS-induced DNA damage, genomic vulnerability of hepatocytes and T-lymphocyte suppression contribute to HCC development[19].
Also, alcohol catabolism impacts several steps of lipid metabolism, which leads to liver steatosis and inhibition of fatty acid oxidation[29].
Reversibility of gene expression via epigenetic alteration is an important biological phenomenon that often plays a role in tumorigenesis. Epigenetic mechanisms affected by excessive alcohol consumption lead to altered DNA methylation and acetylation.For instance, altered acetylation is associated with hepatic steatosis alcohol-induced HCC[29]. Overexpression of c-Met and hepatocyte growth factor is directly associated with promoter hypomethylation in circulating tumor cells (CTCs) of HCC in a syngeneic BALB/c mouse tumor model[30].
Moreover, alcohol abuse is associated with HCC via impaired metabolism, such as accumulation of acetaldehyde, hypomethylation, lack of antioxidants and retinoic acid, together with inflammation, oxidative stress, hypoxia and genetic instability[28].
Other mechanisms of progression to cirrhosis and HCC
In addition to the role of viral hepatitis and alcohol in the development of HCC, other possible risk factors include genetic predisposition and congenital abnormalities, toxic exposures (aflatoxin or arsenic contaminated food), and autoimmune diseases of the liver.
Several congenital abnormalities have been shown to predispose patients to liver cirrhosis and HCC. These include hereditary tyrosinemia, Wilson's disease, alpha-1-antitrypsin deficiency, and hemochromatosis[31].
The pathogenesis of aflatoxin B1 (AFB1) - induced HCC includes several mechanisms, including the formation of mutagenic and carcinogenic intermediates and adducts. Aflatoxins are released from food contaminated by the fungi, Aspergillus flavus and Aspergillus parasiticus. A series of chemical transformations occur that result in the conversion of AFB1 to established mutagenic or carcinogenic compounds:aflatoxin-B1 → aflatoxin B1-8,9 exo-epoxide → 8,9-dihydroxy-8-(N7) guanyl-9-hydroxy aflatoxin B1 adduct → aflatoxin B1 formaminopyrimidine adduct. These adducts and intermediates can also directly induce a mutation at codon 249 of the p53 tumor suppressor gene. This replaces arginine with serine, a change that reverses the tumor suppressing ability of the gene. There are reports that suggest that AFB1 acts synergistically[32]with HBV to induce HCC. Additive interactions have also been reported[33].
In a systematic review, Tansel et al[34]demonstrated a relationship between increased risk of developing HCC in patients with liver cirrhosis as a result of autoimmune hepatitis (AIH). The risk of liver cirrhosis from AIH was found to be lower than that of liver cirrhosis secondary to HBV and HCV infection or primary biliary cholangitis. Nevertheless, the risk of liver cirrhosis and HCC from AIH is clinically significant.
ESTABLISHED BIOLOGICAL MECHANISMS OF HCC PROGRESSION
There are several established biological mechanisms involved in the progression of HCC. These include EMT, tumor-stromal interactions, tumor microenvironment,cancer stem cells, and dysregulation of microRNAs and well-known signaling pathways[35,36]. Some of these are discussed below.
EMT
EMT is a biological process that occurs normally during development and wound healing, but is hijacked by cancer cells. During this process, epithelial cells, which are normally attached to a basement membrane and closely adhered to one another, lose their cell adhesive properties and become migratory in nature[37-39]. This endowed mesenchymal behavior permits the successful migration of cells, which if usurped by cancer cells, can promote their dissemination and spread throughout the body.
EMT has been recognized by many in the field to be important for cancer progression[40,41]. In HCC, there have been several reports of EMT effectors such as cadherins, fibronectin, vimentin, and integrins, being altered to permit a more mesenchymal phenotype. Furthermore, transcription factors promoting EMT,including Snail, Slug, Twist and Zeb, are also upregulated during HCC progression[42,43]. Additionally, there have been a number of studies on exosomes,microRNAs, long noncoding RNAs, and regulatory signaling pathways that have been associated with EMT and demonstrate consequences in HCC progression[30,41,44-49].This is indicative of the important role that EMT plays in HCC progression. The molecular mechanisms of EMT may have diagnostic, prognostic, and therapeutic implications in HCC.
Tumor-stromal interactions and role of the tumor microenvironment
Metastasis is the most common cause of cancer-related deaths[30,50]. Worldwide, HCC is a leading cause of death from cancer[51]. However, the molecular mechanisms of HCC and metastasis are still being clarified[50].
Tumor development and malignant progression can be promoted by a constantly changing extracellular environment that is impacted by microenvironmental stimuli,immune cell cooperation, and inflammatory signals. There is communication between hepatic tumor cells and non-tumor stroma. The non-tumor stroma consists of components of the extracellular matrix (ECM) such as non-malignant fibroblasts,immune and endothelial cells, collectively known as the peri-tumoral microenvironment[52]. Major alterations to the hepatic microenvironment and cells in chronic liver disease influence cancer development[53]. For example, a hypoxic microenvironment in primary HCC is strongly associated with progression and angiogenesis. The consequent enhanced blood supply in the tumor mediates growth formation and metastasis[54].
According to previous studies, tumor cells cross-talk with the abnormal microenvironment, ECM, inflammatory cytokines, chemokines and upregulated growth factors, contributing to increased angiogenesis[55,56]. Although the molecular mechanisms of tumor-stromal interactions are still being clarified, existing evidence show an accumulation of hepatic stellate cells (HSCs), triggered by hypoxia-induced platelet-derived growth factor-BB (PDGF-BB), and proliferation in the tumor stroma,as well as an increase in VEGF-A expression in HSCs leads to HCC angiogenesis[54].
Interactions between normal tumor-suppressive microenvironment and hepatic stellate cells and normal liver fibroblasts have been reported[53]. One of the major factors in liver fibrosis and cirrhosis is activated HSCs[53]. The important paracrine interactions between activated HSCs and hepatocytes impact HCC proliferation and metastasis[57-59]. HSCs (also known as peri-sinusoidal cells), one of the components of the cellular tumor microenvironment in HCC, are responsible for collagen synthesis in the liver[51]. As liver damage occurs, activated HSCs accumulate in the ECM and induce hepatic fibrosis and hepatocarcinogenesis[51].
The exact molecular mechanisms of interactions between non-tumor stromal constituents (specifically macrophages) and hepatic cancer cells are unclear. Studies in mice have shown induced macrophage infiltration of alternatively activated phenotype M2 pro-tumor monocyte-derived macrophages into tumors developed in the chronically damaged livers of mice injected with carbon tetrachloride (CCl4) for 7 weeks[52]. Therefore, an inflamed liver background is favorably associated with increased cancer development[52].
Cancer stem cells in HCC
Liver lineage studies have uncovered four maturational levels of cells that allow the liver to strike a perfect balance between cell gain and cell loss. These include mature hepatocytes, oval cells, bone marrow cells and hepato-pancreas stem cells[60]. These different levels of stem cells integrate to respond to loss of liver cells in the body in several ways, and are thus implicated in liver cirrhosis and HCC.
The cancer stem cell (CSC) theory has been proposed as an explanatory mechanism of HCC metastasis, progression and aggressiveness. CSCs, like regular stem cells,have self-renewing features and are capable of differentiating into tumor cells of varying phenotypes and through several pathways, partly accounting for the heterogeneous clinical presentation of HCC[61]. Previous research has successfully demonstrated that liver cells are directly involved in hepatocarcinogenesis[62], and transformation of these cells may give rise to CSCs. Some reports also suggest that cancer cells in HCC develop from dedifferentiation of mature hepatocytes rather than from uncontrolled proliferation of liver stem cells[63], with intrinsic factors (genetics,autoimmune diseases) contributing, and extrinsic factors (HBV, HCV, alcohol, AFB1)accounting for 70%-90% of the transformation of small hepatocyte-like progenitor cells to cancer cells of HCC[64]. Nevertheless, the correlation between stem-cell division and cancer risk cannot distinguish the effect of intrinsic factors from that of extrinsic factors.
Stem cells originating from the bone marrow, known as bone marrow-derived stem cells, have been demonstrated to be involved in the progression of HCC. Yavorkovsky et al[65]observed the biomarkers when liver trauma simulating HCC was induced with allyl alcohol and demonstrated that only bone marrow-derived stem cells were activated to respond to the trauma.
Stem cells originating from the canal of Hering (oval cells) are mobilized in chronic liver injury[66]. Oval cell biomarkers include γ-glutamyl transpeptidase, glutathione-S-transferase, OV6, α-fetoprotein, neural cell adhesion molecule 1, and chromogranin A[67]. The normal compensatory mechanisms that mobilize stem cells during liver injury are altered in HCC in such a way that promotes progression of the carcinogenic process.
Various models are being used to explain cancer development and intra-tumoral heterogeneity in HCC. These include CSCs, cancer cell plasticity and the clonal evolution model, to mention a few[68]. While the majority of heterogeneous tumor cells stay inactive[69], a small subgroup comprised of CSCs and cancer initiating cells,facilitate tumor development and growth[70-72]. Phenotypic plasticity of cancer cells,which allows conversion from cancer stem cell to non-CSC and vice versa, is one of the proposed mechanisms that may be responsible for the intra-tumoral heterogeneity found in solid tumors[73]. According to previous studies, underlying molecular mechanisms of EMT and CSCs were found to be associated with a high risk for poor prognosis of cancer patients[68].
During normal development, EMT plays a crucial role in organogenesis[74]. At the time of early embryogenesis, through EMT, cell-cell adhesive epithelial cells undergo trans-differentiation and become mobile mesenchymal cells that can migrate, and invade into neighboring tissues and have increased resistance to apoptosis[73-75]. On the other hand, mesenchymal cells can transform back to epithelial cells via the process of mesenchymal-to-epithelial transition, or MET. These reprogramming processes emphasize the epithelial cell plasticity[73]facilitating metastasis to distant and local anatomical sites via increased invasive and migratory functions[68,73,74].
The CSC hypothesis in cancer remains controversial. While some studies have demonstrated the CSC hypothesis in brain, skin, and colon cancers, others have suggested that tumor-initiating cells (TICs, CSC-like cells) exist instead of CSCs in other cancer types[69,76]. Some studies have demonstrated that HCC arises from either TICs or hepatocytes. According to previous research based on drug-treated HCC patients, TICs are the main trigger of tumor development and progression[61].However, the exact origin of TICs is still not completely understood.
Liver CSCs (LCSCs) have many analogous characteristics to normal liver stem/progenitor cells. In addition to self-renewal and tumorigenesis abilities, LCSCs have been implicated in therapeutic drug resistance and relapse in patients[77]. Longterm inflammatory microenvironment, caused by HBV or HCV, chronic alcohol consumption or NASH, and progression of HCC[35]highly contribute to reprogramming of non-CSC into CSCs[78]and the acquisition of CSC-like properties by non-CSCs through carcinogenic dedifferentiation[79].
Identification of tumor - specific biomarkers and discovery of molecular mechanisms are crucial to establish effective therapeutic and early detection strategies for cancer[60,80]. Through the work of several investigators, we are now familiar with some of the putative surface markers for liver CSCs, including epithelial cell adhesion molecule (EpCAM)[81], CD90[82], CD133[83], CD44[84], and CD13[85]. However, there is still uncertainty as to which cell surface markers best identify CSCs in different cancers.
Therapeutic approaches involving inhibitor targeting of signaling pathways, such as Wnt, hedgehog (Hh), TGF-β and Notch signaling, have been shown to diminish LCSC self-reprogramming, metastasis and tumor proliferation[60]. Moreover, drugs designed to modulate cross-talk between CSCs and cancer cells and the tumor microenvironment may have success in inhibiting tumor growth[60]. Other efforts to target LCSC markers and epigenetic modulators could produce promising results.
OTHER MECHANISMS OF HCC PROGRESSION
Another established mechanism of HCC progression is senescence bypass. The liver cells have powerful regenerative abilities. Progenitor cells rapidly divide to restore the balance offset by tissue loss. However, these cells reach a Hayflick limit, a point where cell division is permanently arrested after a number of divisions. The cells are said to exhibit replicative senescence. Replicative senescence can be due to (1) shortening of telomeres in the absence of telomerase, thereby halting cell division; (2) telomericindependent oncogene activation; and (3) elevated ROS. Telomere shortening triggers the DNA damage response, which is thought to activate several signaling pathways,including the p53-p21pRB pathway, bringing replication to a halt. Non-telomeric senescence utilizes both ATM/Chk/p53 and p16-pRB pathways. Oncogene-induced senescence is closely associated with DNA hyper-replication that succeeds oncogenic activation. Several oncogenic pathways have been reported to be involved in triggering oncogene-induced senescence, including activated Ras, c-myc or Wnt/βcatenin[86,87]. Given the tumor suppressing tendency of cell senescence, bypassing it can result in the proliferation of genetically mutated cells, further DNA instability and propagation of HCC. Researchers have been exploring cell senescence induction as a potential strategy in cancer therapeutics.
Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) plays a critical role in how cells respond to stressful stimuli, including infections and ultraviolet radiation[88]. Inflammatory responses mediated by the NF-kB signaling pathway have been reported to be involved in perpetuating the malignant state. NF-kB activation suppresses apoptosis[89], activates EMT[90], represses maspin (a metastasis suppressor gene)[91], and targets VEGF and other angiogenic factors required in forming new blood vessels that supply HCC[90].
There has been considerable advancement in understanding the fundamental epigenetic mechanisms in gene expression, which is now allowing for the development of novel insights into chronic liver disease epigenetic control[92]. For example, loss of DNA methylation has been pointed to as potential diagnostic markers in HCC progression. Some studies have also suggested that non-coding RNAs (ncRNAs) such as microRNAs (miRNAs), small non-coding RNAs (sncRNAs),long non-coding RNAs (lncRNAs), RNA interference (RNAi), small interfering RNAs(siRNAs), and piwi-interacting RNAs (piRNAs), could serve as therapeutic strategies for HCC[93,94]. Several preclinical studies have shown that significant tumor suppression can be achieved by modulating ncRNAs[93,94].
Other interesting factors that have been shown to correlate with HCC patient prognosis are molecular stratification and mutational signatures[95,96]. There are different classes of liver cancer based on varying molecular features and cell of origin[96]. It has been shown that each stratification has a different implication on patient prognosis[95-97]. For example, proliferative subclasses result in a more aggressive phenotype and poorer patient outcomes[97].
In terms of mutational signature, there are several genetic alterations that are promising for therapeutic interventions. For instance, approximately 15% of HCCs harbor amplifications at 11q13 and 6p21[95]. Currently, a better understanding of how molecular stratification and mutational signatures affect HCC progression is still needed before they can be used as therapeutic strategies or biomarkers in a clinical setting.
CTCs in HCC
There is increasing evidence that CTCs play an important role in HCC progression.CTCs are considered an intermediate stage of metastasis. They are cancer cells that have dissociated from the primary tumor, enter circulation, and may subsequently form metastatic lesions[98,99]. There is strong interest in studying CTC biology to understand their molecular mechanisms and how they affect metastasis. Moreover,CTCs have clinical applications, such as diagnostic applications circumventing the need for invasive tissue biopsies[100].
As illustrated in Figure 2, a considerable amount of data has been and can be gathered through the study of CTCs in HCC. Through isolation, characterization and correlation of CTCs with pathological features, as well as disease stage, researchers have shown that a greater CTC count in patient blood is associated with poorer HCC prognosis[53,101-106]. As there is a current lack of reliable biomarkers for the early, noninvasive detection of HCC, a few studies have demonstrated the potential feasibility of using CTCs as a possible diagnostic marker[104,107-110]. Although CTCs are found in very low numbers in the blood[110,111], the advent of new single-cell sequencing technologies and methods to successfully expand CTCs in long-term cultures has enabled their molecular profiling and characterization[104,110,112-115], hence making CTCs promising diagnostic biomarkers in HCC.
IMMUNOMODULATORY MECHANISMS IN HCC
Additionally, several immune mechanisms have been observed to be dysregulated during HCC progression[116]. For instance, HCC is a cancer arising against the backdrop of an inflammatory state in the liver. HBV, HCV, and many of the other etiological factors discussed earlier in this article give rise to chronic inflammation. In turn, this leads to the production of inhibitory cytokines such as IL-10 and transforming growth factor beta (TGFβ), which dampen the immune response and favor tumor growth[117-119]. During HCC progression, regulatory T cells and myeloidderived suppressor cells are also recruited to the tumor site as a result of these cytokine secretions, adding to the already immunosuppressive environment[116,118,120].Lastly, it has been found that several checkpoint inhibitor receptors such as CTLA4 and PD-1 are commonly upregulated in immune cells in the HCC setting. With more checkpoint inhibitor receptors being expressed on these immune cells, they are unable to become active and counterattack tumor cells for clearance from the body[119,121,122].
Ironically, a fundamental characteristic of the liver may also permit tumorigenesis.The liver is immunologically tolerant. This is because the liver is in constant contact with microbiota from the gut and therefore needs to have a tolerant immune response so that it does not become hyperactivated[116,119]. This, in conjunction with the supplementary immunosuppressive mechanisms that develop during HCC progression, enable tumors to grow. This irony makes exploration of immunotherapy for HCC a challenging but potentially exciting prospect to consider.
Indeed, several studies have shown that immune checkpoint inhibitors have had some efficacy in preclinical and early stage clinical trials of HCC. Additionally, the fact that sorafenib, the current first line treatment for advanced HCC, has been noted to exhibit some immunomodulatory effects, seems to suggest the potential efficacy of immunotherapeutic strategies in HCC[122-125].
NEURAL REGULATION OF HCC
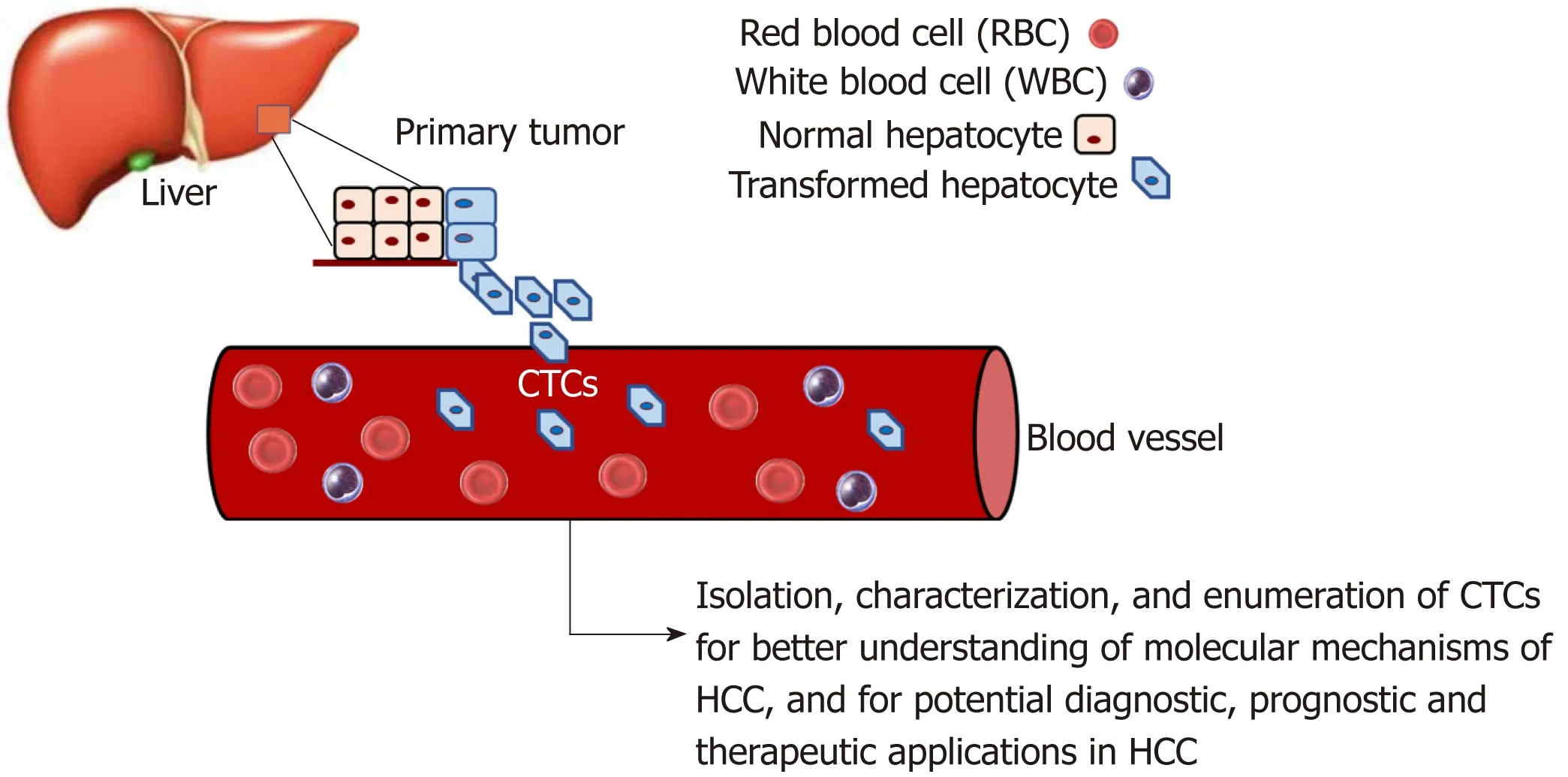
Figure 2 Use of circulating tumor cells as a non-invasive means to study HCC progression.
Tumor cells and the cells in the tumor microenvironment are affected by stress physiology[126]. Neuroeffector molecules can reach the tumor microenvironment via the circulatory system or nerve fibers. During threatening or stressful life circumstances, there is an activation of the sympathetic nervous system, which mediates fightorflight stress responses. The hypothalamus-pituitary-adrenal axis is responsible for mediating withdrawal responses from more profound and overwhelming threats. The neurotransmitter norepinephrine is released by the sympathetic nervous system nerve fibers, while the major stress hormone cortisol is released into the blood by the adrenal gland upon hypothalamus-pituitary-adrenal activation[126]. Cortisol is secreted by the adrenal glands. However, its secretion is regulated by the pituitary gland. Under conditions of severe psychological stress,corticotropin-releasing factor upregulates the secretion of adrenocorticotropic hormone by the pituitary gland. The adrenocorticotropic hormone in turn upregulates the secretion of cortisol[127]. Cortisol can reach the tumor microenvironment via circulating blood, while norepinephrine can do so by being released from nerve fibers(carried by blood vessels), which are recruited in larger amounts by some tumors when these tumors secrete nerve growth factors. Cortisol and norepinephrine binding to the intracellular glucocorticoid receptor (located within the cell) or the beta adrenergic receptor (located on the cell surface) can trigger cellular responses[126].
It has long been recognized that psychosocial conditions affect the progression of some cancers[126]. In fact, epidemiological studies have shown that there is an accelerated progression of various cancers among patients with high stress levels or low social support[126]. While the relationship between stress and cancer development is not fully understood, some studies have shown that psychological stress causes abnormal immune responses, which are associated with cancer pathogenesis[128,129].Cortisol release has been linked to the development and progression of, and survival from various cancers[130-134]. Cortisol inhibits immune responses, which allow cancer cells to evade the immune system[127,134].
Prostate cancer patients have also been shown to have high cortisol levels compared to low risk individuals[131], and breast cancer patients were reported to have high serum cortisol levels, which can be downregulated by emotional support[135].
Serum levels of cortisol have been shown to be higher in HCC patients than in healthy individuals[134]. Studies by Wu and colleagues have shown that exposing HCC cell cultures to cortisol represses p53 expression by upregulating expression of the p53 suppressor Bcl2L12. This suggests that cortisol is a factor that plays a role in the development of HCC[134]. Consequently, it has been suggested that cortisol may be a therapeutic target in HCC treatment[134].
Oxytocin is a neuropeptide hormone produced by hypothalamic neurons and has multiple roles in the central nervous system. While oxytocin is best known for its role in the female reproductive system (milk ejection), further research has shown that oxytocin also plays important roles in complex social behaviors, including stress and trust, anxiety, social interaction and bonding, and parental care, as well as in neuropsychiatric disorders linked to such social behaviors[136,137]. Oxytocin and its receptor have more recently been shown to play roles in some cancers[138-142].
Cortisol has also been linked to some functions of oxytocin[140]. Some studies have shown that higher oxytocin levels and increased social support (a known prognostic player in cancer) are associated with diminished effects of stress. In a study by Mankorious and colleagues, it was shown that there is a cross-talk network between oxytocin and cortisol at the molecular level, where the carcinogenic effect of cortisol was reversed by oxytocin via autophagy in human ovarian cancer cells in vitro[140].
It is known that the effects of oxytocin in cancer may depend on cell type, hormone concentration, its interactions with other hormones in the microenvironment, and the location of its receptor on the cell membrane[137]. Unpublished work from our laboratory analyzing data from sequenced HCC and pancreatic cancer cases in the TCGA dataset showed that genetic alterations in the oxytocin and oxytocin receptor genes were associated with lower median months of overall survival. It would be interesting to determine whether there could be an interaction between oxytocin and cortisol, which could be involved in a potential neural regulation of HCC as well as other gastrointestinal cancers.
CONCLUSION
There has been minimal improvement in the prognosis for HCC patients over the past two decades. The detailed molecular mechanisms of HCC progression remain unclear, and there is an urgent need to better understand the mechanisms underlying HCC progression so as to develop novel and effective therapeutic strategies and reliable prognostic biomarkers. Further, a better understanding of mechanisms of HCC development can further aid efforts at developing effective preventative strategies. This review provides a summary of some of the mechanisms of HCC etiology, and some of the well-established as well as a few recently proposed mechanisms of HCC progression.
杂志排行

World Journal of Gastroenterology的其它文章
- Cyst fluid glucose: An alternative to carcinoembryonic antigen for pancreatic mucinous cysts
- Congenital peritoneal encapsulation: A review and novel classification system
- Autoimmune hepatitis and IgG4-related disease
- Electroacupuncture at ST36 modulates gastric motility via vagovagal and sympathetic reflexes in rats
- Role of D2 gastrectomy in gastric cancer with clinical para-aortic lymph node metastasis
- Risk factors for progression to acute-on-chronic liver failure during severe acute exacerbation of chronic hepatitis Β virus infection