亲水作用色谱-串联质谱法测定蜂蜜中链霉素和双氢链霉素残留
2019-06-24魏莉莉刘艳明孙立臻宿书芳山东省食品药品检验研究院山东济南250101
魏莉莉, 薛 霞, 刘艳明, 孙立臻, 程 志, 宿书芳, 赵 寅(山东省食品药品检验研究院,山东 济南 250101)
链霉素(streptomycin,STR)和双氢链霉素(dihydrostreptomycin,DHSTR)属于氨基糖苷类抗生素,对革兰氏阴性菌有明显的抗菌活性效果[1],可以预防多种动物疾病。链霉素和双氢链霉素能够有效地治疗蜜蜂的腐蛆病[2],在养蜂行业应用普遍,但由于管理和使用的不科学,常造成蜂产品中该类物质的残留。长期食用链霉素和双氢链霉素超标的蜂产品,会对健康产生一定的危害,尤其是听觉神经[3]。因此,国内和国际上对蜂产品中链霉素和双氢链霉素的限量值均有相关规定。我国《绿色食品蜂产品》(NY/T 752-2003)中规定了链霉素的最大残留限量为20 μg/kg;德国规定蜂蜜中链霉素的限量为20 μg/kg。因此,建立蜂蜜中链霉素、双氢链霉素残留量的先进、高效、准确的检测方法对维护人类的饮食健康具有重要意义。
目前报道的链霉素和双氢链霉素的检测方法主要有酶联免疫法[4]、液相色谱法[5-7]和液相色谱-串联质谱法[8-16]。酶联免疫法操作相对简单,但是测定结果的重现性差,准确度低,易造成结果的假阳性或假阴性,常用来进行初筛检测。链霉素和双氢链霉素极性大,分子结构中缺少发色基团和荧光基团,采用液相色谱法测定时,往往需要对化合物进行柱前或柱后衍生[6]。该方法操作繁琐,衍生条件要严格把控,而且无法有效分离链霉素和双氢链霉素,因此不能适应残留检测要求。液相色谱-串联质谱法在抗基质干扰能力、准确定性和灵敏度等方面具有较强的优势,目前在兽药残留检测方面的应用越来越广泛。现有标准方法[9,10]中前处理技术主要采用双柱串联测定蜂产品中的链霉素和双氢链霉素,方法复杂,检验成本高;为了增加链霉素和双氢链霉素的色谱保留行为,在洗脱液或流动相中加入庚烷磺酸钠等离子对试剂,易污染色谱柱和质谱检测器。因此,净化方式和色谱保留行为是链霉素和双氢链霉素测定的难点。
HLB固相萃取柱是一种通用型的固相萃取柱,在净化糖类、蛋白质、脂肪等方面有优异的性能,常被用作测定食品中链霉素和双氢链霉素的净化方式之一,但是需要在提取液中加入离子对试剂才能保留。
本研究采用三氯乙酸(TCA)缓冲溶液提取蜂蜜中的链霉素和双氢链霉素,采用HLB小柱净化,经亲水作用Obelisc R色谱柱分离,MS/MS检测,避免了离子对试剂的引入,取得了满意的结果。
1 实验部分
1.1 仪器、试剂与材料
Triple Quad 5500超高效液相色谱-串联质谱仪(配有电喷雾电离(ESI)源)(美国AB Sciex公司);3-18K台式高速离心机(德国Sigma公司);BT 125D电子天平(德国Sartorius公司);MS3型旋涡混合器(德国IKA公司);SB-800DTD超声清洗仪(宁波新芝生物科技股份有限公司);Milli-Q超纯水系统(美国Millipore公司)。
甲醇、乙腈、甲酸(色谱纯,德国Merck公司);三氯乙酸、盐酸、氢氧化钠、氨水、十二水合磷酸氢二钠(分析纯,国药集团化学试剂有限公司);链霉素标准品(CAS号:3810-74-0,纯度96.4%)和双氢链霉素标准品(CAS号:5490-27-7,纯度94.4%)均购于德国Dr.Ehrenstorfer公司;Oasis HLB固相萃取柱(60 mg/3 mL,美国Waters公司);实验用水为超纯水;实验用蜂蜜均购自山东省各大超市。
1.2 标准溶液的配制
标准储备液:准确称取适量链霉素、双氢链霉素标准品,置于10 mL容量瓶中,用水溶解至刻度,配制成质量浓度为1 g/L的标准储备液,于-18 ℃避光保存。
混合标准工作液:将标准储备溶液用水逐级稀释,配成质量浓度为1 mg/L的混合标准工作液。
1.3 样品的制备
无结晶的实验室样品:搅拌均匀;有结晶的样品:在密闭情况下,置于不超过60 ℃的水浴中温热,振荡,待样品全部溶化后搅拌均匀,迅速冷却至室温。
1.4 样品前处理
1.4.1提取
准确称取5 g试样(精确至0.01 g),置于25 mL比色管中,加入10 mL 20 g/L三氯乙酸水溶液(含50 mmol/L磷酸氢二钠,pH 6.8)涡旋混匀,超声15 min,取出后,用提取液定容至25 mL,摇匀,得到试样溶液。将试样溶液转移至50 mL具塞离心管中,以8 000 r/min离心5 min,收集上清液待用。若样品中有少量花粉,可用滤纸过滤,收集滤液待用。
1.4.2净化
依次用3 mL甲醇、3 mL水活化Oasis HLB固相萃取柱。准确移取5 mL上清液,以不高于1.0 mL/min的流速全部通过固相萃取柱,弃去滤液;用4 mL超纯水分两次淋洗,弃去淋洗液,将小柱抽干;用1 mL甲酸-乙腈-水(2∶5∶93,v/v/v)洗脱溶液洗脱并收集洗脱液,抽干固相萃取柱。向收集的洗脱液中加入1 mL氨水-乙腈(2∶98,v/v),涡旋混匀后,经微孔滤膜过滤,供液相色谱-串联质谱仪分析测定。
基质标准工作溶液:取6份空白样品(5.00±0.02)g,置于25 mL比色管中,按1.4.1节和1.4.2节操作制得空白提取液。分别吸取2.5、5、10、25、50和100 μL混合标准工作液,用空白提取液定容至1.0 mL,配成质量浓度为2.5、5、10、25、50和100 μg/L系列基质标准工作液。

表2 采用4种小柱净化蜂蜜样品时STR和DHSTR的回收率Table 2 Recoveries of STR and DHSTR in honey samples cleaned up by four kinds of SPE column
1.5 分析条件
1.5.1色谱条件
色谱柱:SIELC Obelisc R柱(150 mm×2.1 mm,5 μm);柱温:40 ℃;流动相:A相为乙腈溶液,B相为0.5%(v/v)甲酸水溶液;流速:0.3 mL/min。梯度洗脱程序:0~0.5 min,75%A;0.5~3.8 min,75%A~45%A;3.8~5.0 min,45%A;5.0~6.0 min,45%A~10%A;6.0~8.5 min,10%A,8.5~8.6 min,10%A~75%A;8.6~14.0 min,75%A;进样量:10 μL。
1.5.2质谱条件
离子源:ESI源;离子源温度:550 ℃;检测方式:多反应监测(MRM)模式;扫描方式:正离子模式扫描;电喷雾电压:5 500 V;雾化器压力:345 kPa;气帘气压力:138 kPa;辅助气压力:345 kPa;碰撞气压力:55 kPa;雾化气、辅助气:零级空气;气帘气、碰撞气:高纯氮气。
链霉素和双氢链霉素的定性离子对、定量离子对、采集时间、去簇电压及碰撞能量见表1。

表1 链霉素和双氢链霉素的质谱参数Table 1 MS/MS parameters of streptomycin (STR)and dihydrostreptomycin (DHSTR)
* Quantitative ion.
2 结果与讨论
2.1 前处理方法的优化
2.1.1净化方式的选择
蜂蜜样品含有大量的果糖和葡萄糖,为了达到去除杂质的目的,需要在前处理过程中对目标物进行净化、富集。固相萃取法由于简单、快速、高效等特点被广泛应用于蜂蜜中链霉素和双氢链霉素的净化。本实验比较了Waters Oasis HLB(60 mg/3 mL)、PRiME HLB(60 mg/3 mL)、MCX(60 mg/3 mL)和WCX(60 mg/3 mL)4种固相萃取柱。4种小柱的净化方法及链霉素和双氢链霉素的回收率见表2。结果表明,采用MCX小柱时目标化合物的回收率最低,主要是因为MCX小柱是混合型强阳离子交换柱,与链霉素和双氢链霉素的氨基作用力太强,只用氨水甲醇很难洗脱,文献[10]报道洗脱需要用30 mL磷酸盐缓冲液,但是对后续浓缩造成一定的困难;WCX柱是弱阳离子交换柱,与目标物的氨基作用力相对较弱,回收率有所提高,但是仍不到80%;PRiME HLB柱是一种通过型的固相萃取柱,在净化磷脂、蛋白质等杂质上有出色的性能,但在净化糖类物质方面效果不理想;Oasis HLB柱在去除糖类、蛋白质等杂质上有一定的优势,虽不能直接保留目标物,但是借助一定的提取溶剂,两种化合物均能得到很好地保留,回收率在90%以上。因此,本研究采用Waters Oasis HLB柱作为净化柱。

图1 不同提取剂对链霉素和双氢链霉素回收率的影响Fig.1 Effect of different extraction solvents on the recoveries of STR and DHSTR Solvent 1:water;solvent 2:50 mmol/L phosphate buffer solution (pH 6.8);solvent 3:20 g/L trichloroacetic acid aqueous solution (including 50 mmol/L phosphate,pH 6.8);solvent 4:50 mmol/L 1-heptanesulfonic acid sodium buffer solution.
2.1.2提取溶剂的选择
链霉素和双氢链霉素属于碱性化合物,易溶于水,难溶于甲醇、乙腈等有机溶剂,因此常采用缓冲液进行提取[9,10]。食品基质中链霉素和双氢链霉素的提取溶剂有三氯乙酸溶液、磷酸盐溶液、乙二胺四乙酸二钠(EDTA)缓冲溶液等。本实验考察了水(提取液1)、50 mmol/L磷酸盐缓冲液(pH 6.8)(提取液2)、20 g/L三氯乙酸水溶液(含50 mmol/L磷酸盐,pH 6.8)(提取液3)和50 mmol/L庚烷磺酸钠缓冲液[6](提取液4)4种提取溶剂的提取效率(见图1)。用提取液1和提取液2提取后,链霉素和双氢链霉素几乎无回收,这主要归因于目标物的极性大,在HLB小柱上基本上无保留。提取液中加入离子对试剂(提取液4)后,目标物与离子对试剂结合,极性变弱,在HLB柱上的保留变强,回收率达到90%以上,但是离子对试剂对色谱柱和质谱检测器会有一定程度的污染。提取液3中加入了三氯乙酸,控制溶液的pH值,回收率也能达到90%以上,作用的原理类似于阳离子交换反相吸附原理。在中性条件下三氯乙酸与目标物的氨基作用,目标物得到保留,增大酸度条件,两者之间的作用力减弱,被洗脱下来。另一方面,三氯乙酸在一定程度上也可起到沉淀蛋白质的作用。因此选择三氯乙酸作为提取剂。
提取剂中三氯乙酸的浓度和提取液的pH值对链霉素和双氢链霉素回收率的影响见图2。当提取剂中三氯乙酸的质量浓度为20 g/L时,链霉素和双氢链霉素的回收率均能达到90%以上,之后再增加三氯乙酸的质量浓度,回收率不再明显增加,因此选取三氯乙酸的质量浓度为20 g/L。

图2 (a)三氯乙酸的浓度和(b)pH值对链霉素和双氢链霉素回收率的影响Fig.2 Effect of (a)mass concentrations of trichloroacetic acid and (b)pH values on the recoveries of STR and DHSTR
随着提取液pH值的升高,链霉素和双氢链霉素的回收率逐渐升高,这归因于pH值升高时三氯乙酸与氢离子的作用力逐渐减弱,而与目标物的氨基作用力逐渐增强。当pH值为6~7时回收率较高且比较稳定;当pH值>7时,回收率逐渐下降。因此,提取液的最佳pH值为6~7。
在实际样品测定中,用20 g/L三氯乙酸水溶液(pH 6.8)提取后,不同蜂蜜样品之间回收率差别较大,且回收率偏低。对提取后的样品进行pH值的测定,发现其pH值在3.5~6.2之间,这是引起回收率偏低的一个重要原因。蜂蜜样品含有有机酸,而提取液无缓冲能力,因此样品加入提取液提取后,pH值会发生变化。为解决此问题,在提取液中加入了磷酸盐缓冲液。本实验考察了提取液中加入10、20和50 mmol/L磷酸盐后,上样溶液的pH值和样品的回收率,发现加入10 mmol/L和20 mmol/L磷酸盐时缓冲能力不足,仍有部样品的回收率偏低,而加入50 mmol/L磷酸盐缓冲效果较好,上样溶液的pH值稳定在6.2~6.7之间。综合以上因素,将20 g/L三氯乙酸水溶液(含50 mmol/L磷酸盐,pH 6.8)作为最终的提取溶剂。
2.1.3洗脱溶剂的选择
考察了洗脱液中甲酸、乙腈和水的体积分别为1 mL+5 mL+94 mL、2 mL+5 mL+93 mL和5 mL+5 mL+90 mL时,链霉素和双氢链霉素的回收率。当洗脱液甲酸-乙腈-水的体积分别为1、5和94 mL时,链霉素和双氢链霉素的回收率分别为79.2%和76.8%,回收率偏低;当洗脱液中甲酸-乙腈-水的体积分别为2、5和93 mL时,链霉素和双氢链霉素的回收率分别为93.2%和98.9%,之后再增加甲酸的体积,回收率不再明显增加,因此选择甲酸-乙腈-水(2∶5∶93,v/v/v)为最终的洗脱溶剂。
本研究考察了洗脱溶剂分别为0.5、1.0、2.0和3.0 mL时对链霉素和双氢链霉素的影响。结果表明,当洗脱溶剂为0.5 mL时,链霉素和双氢链霉素均能被完全洗脱,但是相对标准偏差较大;洗脱液用量过多又会导致方法的检出限偏高。故实验选择1.0 mL的洗脱液进行洗脱。

图3 采用不同色谱柱时链霉素和双氢链霉素(100 μg/L)的色谱图Fig.3 Chromatograms of STR and DHSTR (100 μg/L)on different chromatographic columns a.column:SIELC Obelisc R column (150 mm×2.1 mm,5 μm);mobile phases:0.5% (v/v)formic acid aqueous solution and acetonitrile;b.column:Waters BEH Amide (100 mm×2.1 mm,1.7 μm);mobile phases:0.1% (v/v)formic acid aqueous solution containing 5mmol/L ammonium acetate and acetonitrile;c.column:Waters BEH HILIC (100 mm×2.1 mm,1.7 μm);mobile phases:0.1% (v/v)formic acid aqueous solution containing 5 mmol/L ammonium acetate and acetonitrile.
2.1.4稳定性的考察
用洗脱后的基质空白溶液(未添加氨水乙腈)稀释标准溶液,制备50 μg/L的基质标准溶液,分别放置0、6、12、18、24和48 h后测定,与每次临用前稀释的链霉素和双氢链霉素的基质标准溶液(50 μg/L)的响应值进行比较,以两者的比值(f)考察稳定性,f值越小,稳定性越差。
结果表明,随着放置时间的延长,链霉素和双氢链霉素的f值均下降,放置48 h后,链霉素的f值降为0.52,双氢链霉素的f值降为0.61,说明链霉素和双氢链霉素在甲酸-乙腈-水(2∶5∶93,v/v/v)溶液中稳定性较差。为解决这一问题,在洗脱后的基质空白溶液中加入了1 mL氨水-乙腈(2∶98,v/v)溶液,此时溶液的pH值在4.5左右,用此空白溶液稀释的基质标准溶液在48 h内稳定。
2.2 色谱条件的优化
2.2.1色谱柱的选择
链霉素和双氢链霉素具有较强的极性和离子特性,在反相C18色谱柱上不保留,必须在流动相中加入离子对试剂来改善目标化合物在C18色谱柱上的保留行为,但是离子对试剂会对质谱检测器造成污染。
本实验选择了亲水作用色谱柱分析化合物。实验比较了链霉素和双氢链霉素(100 μg/L)在Waters BEH Amide(100 mm×2.1 mm,1.7 μm)、Waters BEH HILIC(100 mm×2.1 mm,1.7 μm)和SIELC Obelisc R (150 mm×2.1 mm,5 μm)3种色谱柱上的保留行为(见图3)。结果表明,在HILIC色谱柱(见图3c)上,目标物的响应值偏低且峰形较差;在Amide色谱柱(见图3b)和Obelisc R(见图3a)色谱柱上,目标物的响应值无明显差别,但是经过Amide色谱柱分离后目标物的峰宽较宽,且峰形拖尾。因此,本实验选择SIELC Obelisc R作为色谱柱。
2.2.2流动相的选择
SIELC Obelisc R色谱柱是在硅胶表面修饰了羧酸类的官能团,醇类会酯化硅胶表面键合的羧酸,影响物质的保留时间与重现性,因此本实验的流动相选用乙腈而不是甲醇。流动相中甲酸的体积分数影响链霉素和双氢链霉素在色谱柱上的保留。本实验考察了0.2%、0.5%和1.0%(v/v)甲酸水溶液分别作为流动相时对目标物的影响。实验发现随着流动相中甲酸体积分数的提高,保留时间逐渐缩短,响应值提高,但是当甲酸的体积分数大于0.5%后,响应值不再显著变化,且长时间使用酸度较强的流动相也会影响色谱柱的使用寿命。综合考虑,选择乙腈-0.5%(v/v)甲酸水作为流动相。
2.3 质谱条件的优化
链霉素和双氢链霉素的结构中含有多个伯胺,具有弱碱性,易被电离成正离子。本实验采用电喷雾电离源,在正离子扫描模式下对化合物进行母离子扫描,获得链霉素分子离子峰的m/z为582,双氢链霉素分子离子峰的m/z为584;然后以各自的分子离子峰为母离子,对子离子进行二级质谱扫描,选取丰度较强、干扰较小的离子为定量和定性离子。在MRM模式下,优化质谱参数,优化后的质谱条件见表1。

表3 链霉素和双氢链霉素的线性方程、线性范围、相关系数、LOD和LOQTable 3 Linear equations,linear ranges,correlation coefficients (r),LODs and LOQs of the STR and DHSTR
y:peak area;x:mass concentration,μg/L.
2.4 方法学验证
2.4.1线性关系和检出限
对质量浓度分别为2.5、5、10、25、50、100 μg/L的系列基质标准工作溶液进行测定,获得标准溶液的色谱图。以定量离子对的色谱峰面积(y)为纵坐标,以标准工作溶液的质量浓度(x,μg/L)为横坐标,绘制标准曲线,得到线性回归方程。对空白样品加标,以S/N≥3、精密度≤30%为评价依据,确定方法的检出限为2.0 μg/kg;以S/N≥10、精密度≤21%,回收率范围60%~120%为评价依据,确定方法的定量限为5.0 μg/kg。链霉素和双氢链霉素的线性方程、线性范围、相关系数(r)、检出限及定量限见表3。链霉素和双氢链霉素在2.5~100 μg/L范围内,峰面积与质量浓度之间呈良好的线性关系,相关系数均大于0.99,符合方法确证要求。蜂蜜空白样品和加标样品(20 μg/kg)的选择离子流色谱图见图4。
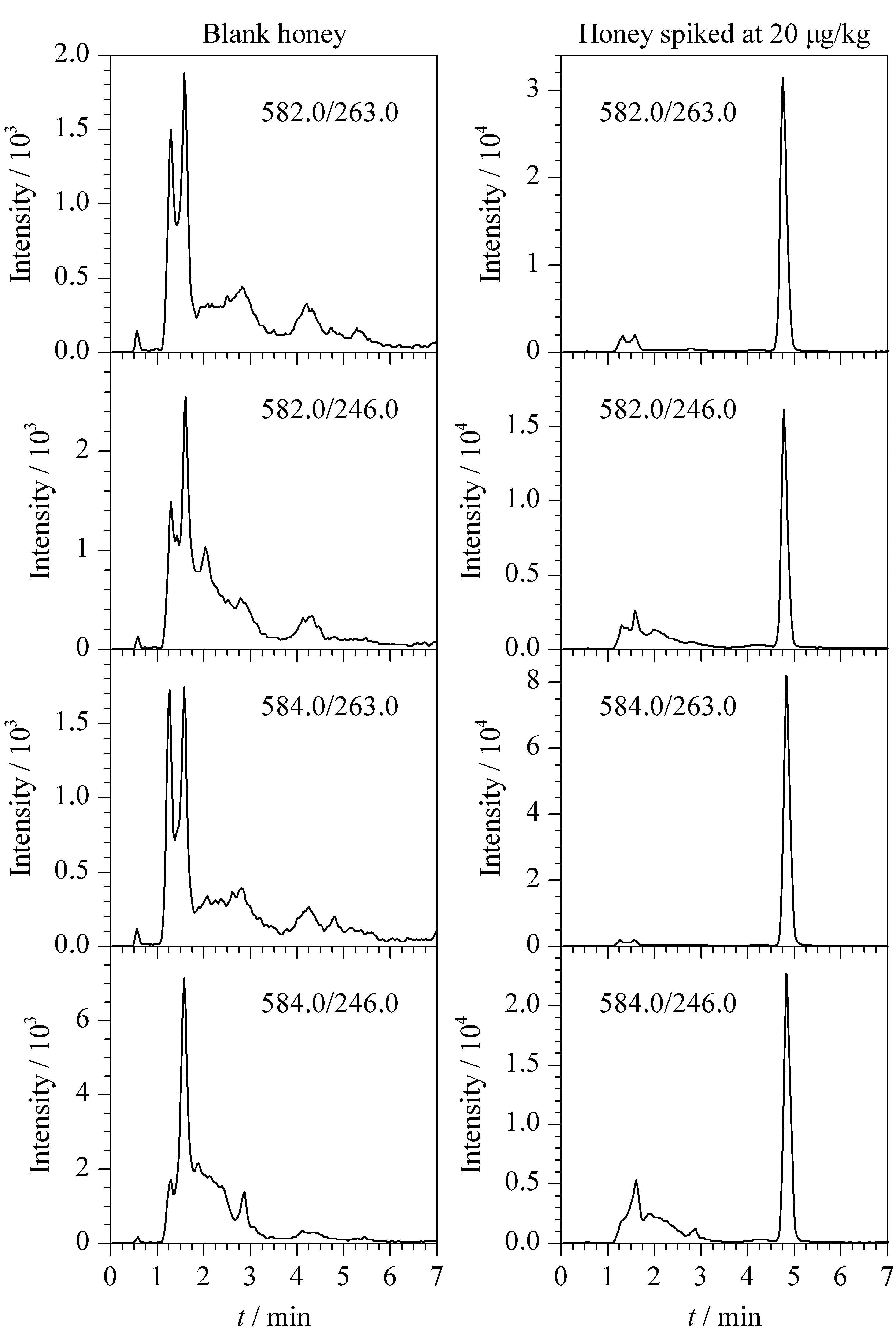
图4 (a)空白蜂蜜和(b)加标蜂蜜(20 μg/kg)样品的选择离子流色谱图Fig.4 Selected ion current chromatograms of (a)blank honey and (b)spiked honey (20 μg/kg)samples
2.4.2回收率与精密度
向空白样品中分别进行低(5.0 μg/kg)、中(20.0 μg/kg)、高(100.0 μg/kg)3个水平的加标回收试验,每种加标样品平行测定6份,考察方法的回收率与精密度。方法的平均回收率为86.9%~113.2%,日内和日间精密度小于10%(见表4)。
2.5 基质效应(ME)
质谱检测常需要通过基质效应评价方法的灵敏度和选择性。ME=空白基质匹配标准溶液的响应值/纯溶剂标准溶液的响应值×100%,若ME>100%,表示基质效应增强;若ME≈100%,表示无明显基质效应;若ME<100%,表明基质效应抑制。本实验中比较了链霉素和双氢链霉素质量浓度同为50 μg/L时的基质效应,链霉素的ME值为90.3%,双氢链霉素的ME值为91.4%。ME值大于90%,表明方法净化效果良好。

表4 链霉素和双氢链霉素的回收率和相对标准偏差(n=6)Table 4 Recoveries and relative standard deviations of the STR and DHSTR (n=6)
2.6 实际样品分析
采用本文建立的方法对山东省各大超市采购的50批次蜂蜜样品进行分析,其中8批次样品中检出链霉素,含量为7.2~76.8 μg/kg,双氢链霉素均未检出。
3 结论
本文建立了亲水作用色谱-串联质谱测定蜂蜜中链霉素和双氢链霉素的分析方法。该方法有以下优点:(1)采用三氯乙酸缓冲溶液提取,HLB固相萃取柱对目标化合物进行净化,可减少基质干扰;(2)亲水作用Obelisc R色谱柱分离目标物,与文献[6,8,9]报道的采用反相色谱柱相比,避免了离子对试剂对色谱柱或质谱检测器的污染;(3)方法的灵敏度高,回收率和精密度均能满足各类检测要求,可应用于蜂蜜中链霉素和双氢链霉素的批量化检验工作的开展。
