基于人皮瓣核酸内切酶抗癌策略的研究进展
2019-06-21王海月曹秀琪
马 龙,王海月,周 津,曹秀琪
(天津科技大学生物工程学院,天津 300457)
恶性肿瘤已成为威胁人类健康的头号杀手.研究[1]估计,2018年全球范围内将会有1810万癌症新增病例和960万癌症死亡病例,并且预计癌症将成为21世纪世界上每个国家人民的主要死亡原因和提高寿命最重要的障碍.因此,研究癌症成因和发展机制,找到高效合理的预防和治疗手段,是医药工作者的重大命题.虽然人类已经取得了一定进展,有不少有效的药物被应用于临床,但是找到新的药物靶点,使药物能够选择性的杀灭癌细胞,降低药物对正常细胞的毒副作用和防止癌症复发,仍是抗癌药物研发所面临的巨大挑战.
不少的抗癌药物都是通过引起过度的 DNA损伤来抑制癌症[2],因此癌细胞已经进化出高效、保守的 DNA修复系统,以防止内源和外源 DNA损伤.然而当特异性 DNA修复酶含量增加时,可以提高肿瘤细胞对抗癌药物的抗性[3],因此人皮瓣内切酶1(human flap endonuclease 1,FEN1)作为一种结构特异型的5′-核酸酶,因其在DNA复制和修复中的重要作用引起了研究者的注意.研究[4]显示,使用 FEN1抑制剂可以直接杀死癌细胞或者增敏某些 DNA损伤剂(DNA damaging agent).FEN1还可以通过涉及“合成致死性”的机制选择性杀死癌细胞.因此,在基于遗传和组合抗癌治疗模式中,开发功能有效的小分子FEN1抑制剂以降低FEN1的活性,在开发靶向抗肿瘤药物中具有重要意义.
1 FEN1的生物学功能和作用机制
FEN1能够特异性地切除在DNA聚合酶催化的DNA链置换合成过程中产生的 5′端单链 DNA或者RNA[5].FEN1是一种结构特异型核酸酶,需要二价金属离子才能发挥催化作用[6].FEN1是真核生物后随链复制(lagging-strand DNA replication)、长补丁碱基切除修复(long-patch base excision repair,LP-BER)以及核苷酸切除修复(nucleotide excision repair,NER)中不可或缺的重要酶[7].另外,FEN1还在维持端粒稳定性、重启停滞的复制叉、DNA碎片化和处理三核苷酸串联重复结构(trinucleotide repeat expansion,TRN)上起重要作用[8].研究[9]表明,FEN1主要有维持端粒稳定、进行碱基和核苷酸切除修复以及促进冈崎片段成熟的功能.在DNA复制和修复中,FEN1和PCNA(proliferating cell nuclear antigen)相互作用形成“滑行夹”,然后去完成核酸切割任务[10]. 在DNA碱基切除修复中,一分子的 FEN1和一分子的APE1(human AP endonuclease)相结合,APE1协同FEN1作用并增强其活性[11].在维持端粒稳定性方面,FEN1和端粒酶形成复合物,并在端粒酶介导的端粒稳定性上发挥作用[12].在酶学机制上,FEN1的底物DNA中未能配对的5′-单链部分,通过一种“杂乱-穿孔-有序”(disorder-thread-order)的机制穿过FEN1的螺旋状门道结构(helical arch).并且 FEN1通过一种“去碱基配对化(unparing)”的机制去定位切割位点,以便进行正确的切割[13-15].底物 DNA 的5′末端的单链部分穿过仅能够容纳单链 DNA的螺旋状门道结构,而其双链部分不能穿过,所以不能接触到酶的活性口袋(图 1[6]).在 5′端的单链区穿过螺旋状门道结构后,FEN1将+1和-1碱基(+1和-1代表 FEN1切割的磷酸二酯键所连接的两个碱基,+1在 5′端,-1在 3′端)和原来互补链的配对碱基分离,伸向酶的活性中心,这样就只有应该被剪切的磷酸二酯键才能够和活性中心的氨基酸残基接触,接近两个二价金属阳离子,以完成DNA切割(图2[6]).这两种机制使得FEN1拥有结构特异性的DNA切割能力,并且发现了活性中心的Y40、R93、R100对FEN1的活性和作用有巨大影响,在底物切割特异性上发挥重要作用.单分子荧光能量共振转移实验(Singlemolecule fluorescence resonance energy transfer)以及生物物理学研究显示,FEN1应用诱导契合模型(induced-fit)来完成底物的特异性选择[16].正是FEN1的这种精准、特异的 DNA底物切割机制保证了基因组的稳定性[17].
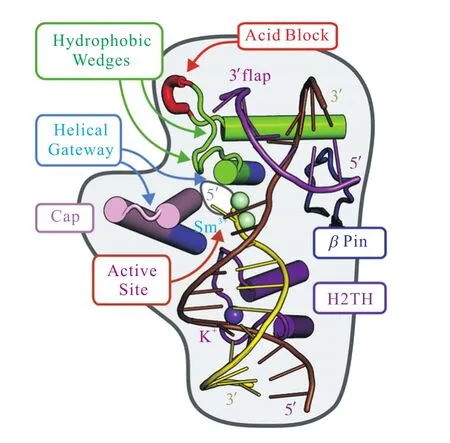
图1FEN1和底物 DNA复合物的晶体结构和结构功能单元Fig. 1 Crystal structure and functional components of FEN1 and substrate DNA complexes

图2 FEN1与底物切割模式Fig. 2 Cleavage pattern of FEN1
2 FEN1在癌症中的作用
鉴于 FEN1在维持基因组完整性方面发挥着至关重要的作用,并且其在多种癌症类型中过度表达,因此它可能作为一种肿瘤治疗靶点[18-19].在 FEN1与肿瘤相关性的方面有不少的报道:(1)其在前列腺癌、胰腺癌、胃癌、肺癌、神经母细胞瘤等肿瘤细胞中高表达[20-23];(2)可作为乳腺癌、卵巢癌、胃癌等肿瘤的生物标志物,而且可以增加肺癌、胃肠癌的易感性[24-28];(3)FEN1的高表达与肿瘤病人的低生存率密切相关,同时还与进行前列腺切除术的前列腺癌病人的不良预后正相关[22,25,29-30];(4)敲低 FEN1有抑制 RAD54B突变的人结直肠癌细胞、抑制人前列腺癌细胞的生长以及增敏曲妥珠单抗的作用,增强胶质瘤细胞对替莫唑胺和顺铂的敏感度;(5)紫杉醇联合FEN1抑制剂对宫颈癌细胞具有协同抗肿瘤作用[31]. 因此,抑制FEN1可起到抑制肿瘤细胞的作用[22,32-37].
3 从合成致死角度探索 FEN1抑制剂抗肿瘤的思路
新的抗肿瘤药物开发原理被不断提出,美国科学家Lee Hartwell和Stephen Friend最早提出了将合成致死性概念用于癌症的治疗[38].所谓合成致死,简单地说就是指在两个非致死基因中,若仅其中的一个基因发生了变异,细胞仍会存活,若两个基因同时发生变异,则将引起细胞死亡,这两个基因则构成合成致死搭档(partner).合成致死策略为寻找抗癌的新靶点提供了全新的思路.癌细胞由于其基因缺陷使它们在合成致死性面前变得更脆弱,这种肿瘤特异性缺陷正好可以被用来研发靶向药物[39-40].比如,发现肿瘤细胞中某个基因失活或者用药物抑制某个基因,与之构成合成致死搭档的另外的一个基因就有可能成为潜在的“靶基因”,那么针对这个“靶基因”的抑制剂就能够特异性的杀死癌细胞,并且不会影响正常细胞的存活(图 3和图 4).总之,合成性致死方法是利用肿瘤细胞和正常细胞间的基因差异来最大化地发挥对癌细胞的杀灭作用,同时将对正常细胞的副作用最小化.因此,合成致死方法针对了不同的基因型,从而可以为病人提供更具个性化的治疗[41],达到“精准”治疗肿瘤的目的[42].

图3 合成致死性原理Fig. 3 Principle of synthetic lethality
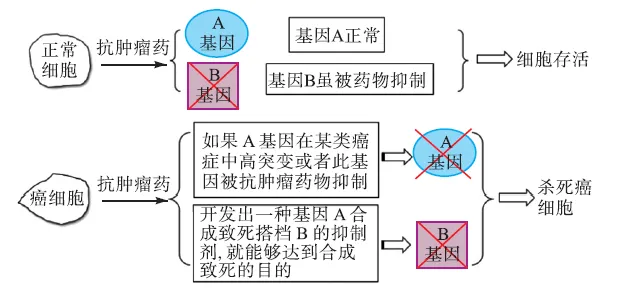
图4 合成致死性方法在抗肿瘤药物研究中可能的应用Fig. 4 Possible applications of synthetic lethality in anticancer drug development
基因组的不稳定性和基因突变被公认为是肿瘤细胞的一大特征[43].DNA损伤及由肿瘤细胞活跃的生长因子和原癌基因诱导的 DNA复制压力造成了基因组的不稳定.DNA损伤修复基因(DNA damage response,DDR)的突变或沉默在肿瘤细胞中很常见,所以 DDR基因也被认为是一类抑癌基因.在肿瘤中,一些 DDR基因的缺失导致其他 DNA修复元素代偿性地上调,这样就会对基于破坏 DNA的放化疗(如 DNA烷化剂等)产生抵抗性,造成治疗效果不佳或耐药性[44-45].因此,癌细胞为了生存而过度依赖的被上调的 DDR通路就可以被作为靶点进行抑制,以引发不可修复的 DNA损伤,达到诱导肿瘤细胞凋亡的目的.简单地说就是 DDR修复的多重机制中潜在地存有多对合成致死搭档[46-47].一个最明显应用合成致死的例子就是在癌细胞中的 BRCA1/2(breast cancer susceptibility gene,BRCA)参与DNA修复,其突变造成 DNA修复障碍,与其协同修复 DNA的聚腺苷二磷酸核糖聚合酶(poly(ADP)ribose polymerase,PARP)旁路则成为癌细胞更为倚重的修复途径.在BRCA1/2突变的癌细胞对 PARP抑制剂的敏感度比非突变细胞大两个数量级[48].在 2016年被 FDA获批上市的 PARP抑制剂 Rucaparib(AG 014699)正是利用了这一原理设计产生的,并且临床试验证实其对铂类药物敏感的高度恶化的浆液性卵巢癌、子宫内膜上皮癌、原发性腹膜癌、输卵管癌和有害生殖系统或体细胞BRCA1/2突变的三阴性乳腺癌等有明显的活性[49-52].有意思的是,尽管 PARP抑制剂最初被设计用来作为抗肿瘤药物的增敏剂,但是有研究显示某些PARP抑制剂(如Olaparib,已由FDA批准)作为单独用药在对癌细胞的抑制作用也非常突出[53].研究发现 FEN1和人体中诸多癌症相关基因如 RAD54B、CDC4、MRE11A、RNF20和 SMC3构成合成致死搭档.其中 RAD54B是一种在体细胞突变成癌细胞的过程中发生变异的基因,具有影响染色体稳定性的作用[32];CDC4是一种在多种肿瘤中突变的基因(如前列腺癌、胰腺癌和结直肠癌);RNF20、SMC3和MRE11A 是在结肠直肠癌中高突变的基因[33].研究表明,FEN1在细胞中可以与多种蛋白交互作用以控制形成信号网络[54],这就为合成致死策略提供了可能.事实上,FEN1被认为可能与DDR通路中的多个重要基因构成合成致死搭档,这些基因的突变可在多种不同肿瘤中出现[55].DNA修复机制与 FEN1抑制剂的作用如图 5所示,其中:BRCA表示乳腺癌易感基因,FANC表示范科尼贫血互补基因,ATM表示毛细血管扩张性共济失调症突变基因,DNA-PK表示DNA依赖性蛋白激酶,XRCC表示X射线修复交叉互补蛋白.
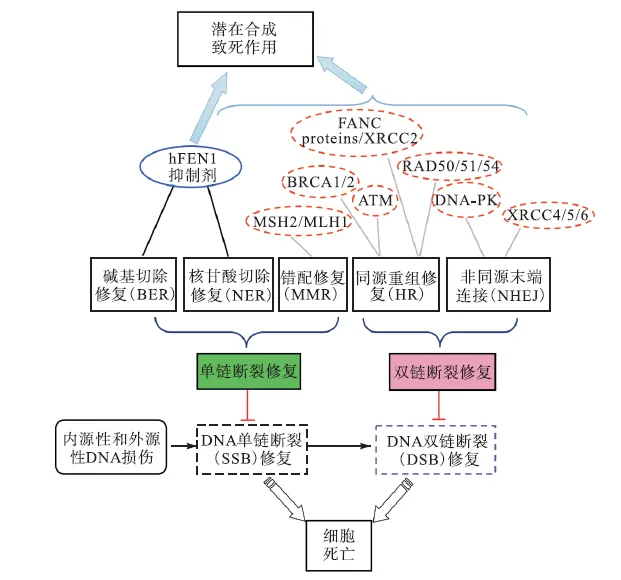
图5 DNA修复机制与FEN1抑制剂的作用Fig. 5 DNA repair mechanism and the roles of FEN1 inhibitors
为了修复内源和外源 DNA损伤,细胞应用一系列DNA修复机制.总体上可以分为DNA单链(DNA single-strand break)和 DNA双链断裂(DNA doublestrand break)修复机制.其中 DNA 单链断裂修复又包括碱基切除修复、核苷酸切除修复、错配修复;DNA双链断裂修复包括同源重组和非同源末端连接.而能够与FEN1抑制剂(蓝色实线椭圆)构成潜在合成致死性搭配的其他 DNA修复系统(红色点线椭圆)的缺失,经常在癌细胞中出现.因此,FEN1的小分子抑制剂是潜在的新型抗癌药物.
4 FEN1抑制剂的研究进展
近年来关于FEN1抑制剂的报道逐渐增多,发现姜黄素能够在蛋白表达水平上下调FEN1的表达,这可能是姜黄素抑制乳腺癌细胞增殖的机制之一[56];还发现姜黄素能够通过对 FEN1的下调增加乳腺癌细胞对顺铂的敏感性[57].中药铁筷子中分离的一种强心苷类成分HTF-1能剂量依赖性地下调FEN1的表达,这也是此化合物能够抑制癌细胞的原因之一[58]. FEN1酶活性抑制剂包括 NSC-281680[34]和NSC-13755[59].NSC-281680能够抑制 FEN1的核酸内切酶活性,进而影响长补丁碱基切除修复.此化合物能够增敏 DNA烷基化抗肿瘤药物替莫唑胺对HCT-116人结肠癌细胞的细胞毒性,为联合用药提供了一种可能[34].通过基于机器学习的虚拟筛选,发现一种名为JFD00950萘醌类化合物具有FEN1催化抑制活性[60].事实上,研究最充分的是基于 N-羟基脲类化合物的抑制剂.这种抑制剂在 2005年被首次报道,在酶水平上其具有纳摩尔级别的 IC50值,其可能的抑制 FEN1酶活的抑制机制为其能够配位结合(coordination)FEN1发挥活性所必需的二价金属镁金属离子,其可能的两种模式如图 6所示[35].在2016年英国谢菲尔德大学的 Jane Grasby教授对这类抑制剂进行了系统的研究,得到了FEN1与化合物1的共结晶,这为研究 N-羟基脲类化合物的 FEN1抑制活性提供了非常重要的依据[61].化合物 1(图 7)能够通过抑制 DNA底物的“去碱基配对化”抑制FEN1的活性,并且化合物 1能够明显增敏 SW620人结肠癌细胞对甲基磺酸甲酯(MMS)的敏感型.此后其他研究组也陆续报道N-羟基脲类化合物能抑制细胞增殖,激发染色体不稳定性,并且能够增敏某些DNA损伤型抗肿瘤药物,这一结论在细胞和动物模型中得到证实[62].
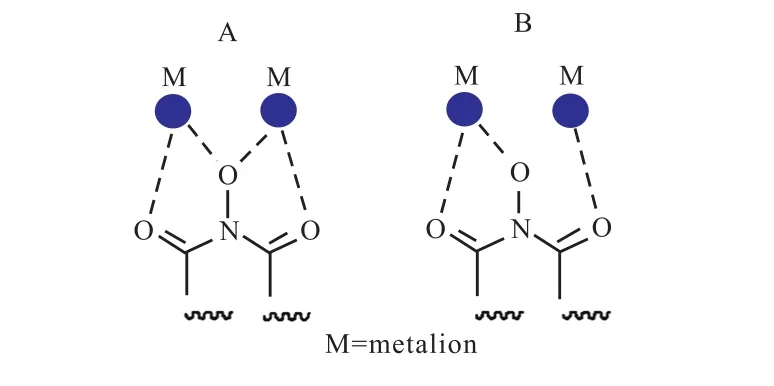
图6 N-羟基脲类化合物的可能作用机制Fig. 6 Possible mechanism of N-hydroxyurea compounds

图7 N-羟基脲类化合物中一种典型代表Fig. 7 A typical representative of N-hydroxyurea compounds
此外,近期报道[23,31,63]还显示,N-羟基脲类化合物还能增敏化疗药物紫杉醇、逆转细胞对顺铂的耐药性以及对 PARP1和 PARP2缺失的细胞表现出高度药物敏感性.
5 结 语
(1) FEN1是一个肿瘤抑制酶,其在维持基因组稳定性方面具有重要作用,但是FEN1的过表达在多种肿瘤中出现,意味着其在肿瘤的快速增殖中扮演重要角色,可以潜在地作为多种肿瘤发生、发展和预后的生物标志物.
(2) FEN1在细胞中蛋白表达水平或催化活性的动态调节过程值得关注.虽然已有一些FEN1抑制剂被发现,但是近年来关于新型的抑制剂报道很少,随着FEN1晶体结构和作用机制的逐步揭示,关于其抑制剂的研究也是今后的一个研究方向.
(3) 随着FEN1和其合成致死搭档的逐步揭示,FEN1抑制剂有可能在治疗基因特异型肿瘤中发挥作用.
