17α-羟化酶缺乏症影像学特征(附1 例报告并文献复习)
2019-06-15刘焦枝湖北医药学院附属襄阳市第一人民医院放射科湖北襄阳441000
宋 萍,王 勇,刘焦枝,杨 峰,李 杨(湖北医药学院附属襄阳市第一人民医院放射科,湖北 襄阳 441000)
病例社会性别女性,27 岁,因四肢无力1 天入院。患者1 天前起床后发现全身无力,表现为抬头困难,四肢无力,不能自行起床,伴有恶心,余无其他不适。既往史:多次晕厥病史,持续约1 min 缓解,无口吐白沫、牙关紧闭现象。无月经来潮,其父母非近亲结婚,家族成员中无类似病史及其他遗传性疾病史。体格检查:体温36.5℃,呼吸23 次/分,脉搏114次/分,血压184/127 mmHg(1 mmHg=0.133 kPa),神志清楚,吐词清楚,颈软,无胡须及喉结,双侧瞳孔等大等圆,直径2.5mm,对光反射灵敏。伸舌居中,口角不歪。双上肢近端肌力2 级,远端肌力4 级,双手握力差,双下肢肌力2 级,四肢肌张力可,四肢浅反射减弱,双侧病理征未引出。专科查体:双乳房未发育,幼女型外生殖器,阴道为盲端,长约4 cm,无阴毛及腋毛,盆腔内空虚未及子宫附件。双侧腹股沟区均可触及光滑活动结节,直径约10 mm。
辅助检查:心电图示:窦性心动过速,缺血型ST-T 改变。血常规:红细胞2.76×1012L-1(偏低);血红蛋白86.0 g/L(偏低),提示贫血。血生化:钾1.53 mmol/L(极低);丙氨酸氨基转移酶149.2 IU/L (偏高);天门冬氨酸氨基转移酶251.3 IU/L(偏高),提示肝功能不全。心肌酶:乳酸脱氢酶693 U/L(偏高);肌酸激酶10 886 U/L (偏高);肌酸激酶MB 同工酶146.70 U/L(偏高);肌红蛋白>1 200.0 ng/mL(偏高),提示有心肌损伤。血总皮质醇:0.20 μg/dL(偏低)。性激素全套:卵泡生成激素(FSH)38.97 mIU/mL(偏低);黄体生成素(LH)14.49 mIU/mL(偏低);催乳素(PRL)17.89 ng/mL;雌二醇(E2)<10.0 pg/mL(偏低);睾酮(T)0.18 ng/mL(偏低);孕酮(P)5.5 ng/mL(偏低)。卧立位肾素-血管紧张素Ⅱ-醛固酮试验:肾素卧位<0.500 0 mIU/L,立位<0.500 0 mIU/L;血管紧张素Ⅱ卧位39.01 pg/mL,立位29.54 pg/mL;血浆醛固酮卧位3.69 ng/dL,立位3.84 ng/dL。ACTH-F 节律(0 时-8 时-16 时):促肾上腺皮质激素(ACTH)8.54-112.85-27.15pmol/L,皮质醇83.46-170.11-106.01 nmol/L,皮质醇节律存在,ACTH 水平显著升高。24 小时尿皮质醇17.33 μg/24 h(偏低)。染色体核型:(46,XY)。
影像表现:肾上腺CT 平扫+增强示:双侧肾上腺体积弥漫性增大,形态失常,其内可见多发结节状及团块状等低混杂密度影(图1~4),边界清楚,CT 值约-65~43 HU,增强后软组织部分延迟期明显强化(图2,3),CT 值约81 HU,脂肪低密度影无明显强化;脾脏体积增大;骨窗示胸腰椎骨质密度普遍减低,上下缘呈双凹状改变(图5)。诊断:①双侧肾上腺增生;②双侧肾上腺多发占位,考虑髓样脂肪瘤可能;③脾大;④胸腰椎骨质疏松。腹部超声示:盆腔内未探及子宫及附件。腹部及盆腔MRI 平扫示:腹盆腔内未见子宫及附件组织(图6,7),双侧腹股沟管近内环处可见直径约10 mm 结节状等T1WI 长T2WI 信号影考虑为发育不良隐睾(图8)。双手X线检查:双手及腕关节组成骨轻度骨质疏松,双侧掌指骨及尺桡骨远端骨骺未闭合(图9)。头部CT 平扫:颅内未见明显异常。垂体MR 平扫未见明显异常。
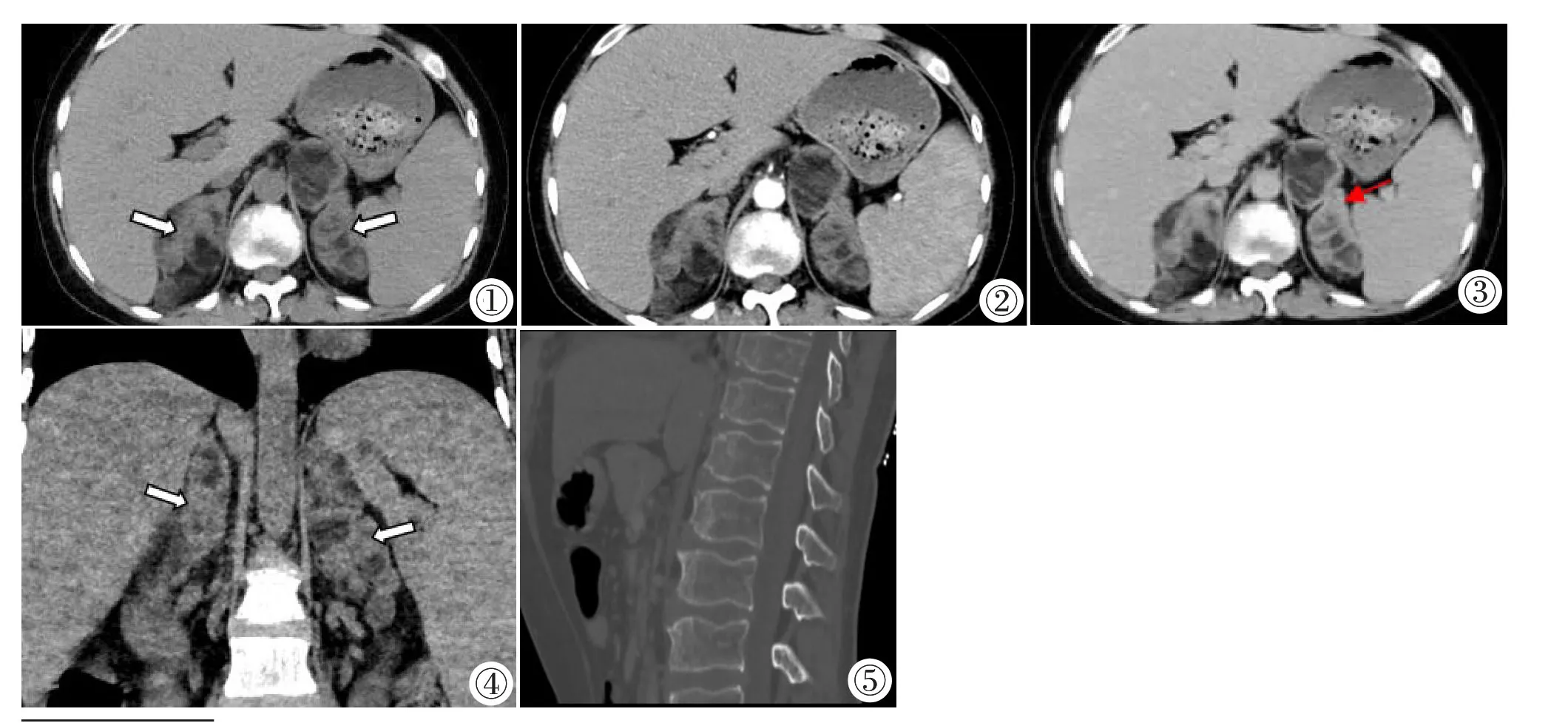
图1~4 肾上腺CT 横轴位平扫及增强 (图1~3)、肾上腺CT 平扫冠状位重建(图4)示:双侧肾上腺多发结节状、团块状软组织密度及脂肪密度影(白箭头),CT 值约-65~43 HU,增强扫描软组织部分延迟期明显强化(红箭头),CT 值约81 HU。图5 骨窗示:胸腰椎骨质疏松。
诊断和治疗:患者为女性外阴,超声及MRI 检查未见子宫及附件,腹股沟区发现发育不良隐睾,染色体核型为46XY,为男性假两性畸形。患者有高血压、低血钾,血皮质醇降低,ACTH 水平显著升高,雌二醇及睾酮水平降低,双侧肾上腺增生。诊断:17α-羟化酶缺乏症。行双侧腹股沟区隐睾切除术,术后证实为睾丸组织。
由于患者社会及心理性别均为女性,性腺及染色体性别为男性,患者最终选择了女性性别。
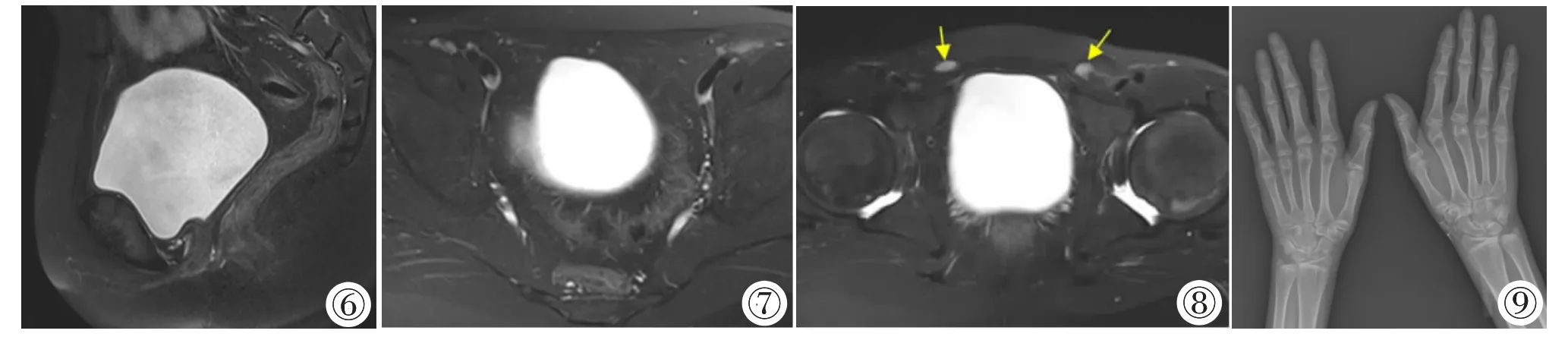
图6~8 盆腔MRI 平扫T2WI 抑脂序列示:女性外阴,直肠前方、膀胱后方无子宫,阴道呈盲端,盆腔内未见附件组织。双侧腹股沟管近内环处可见发育不良隐睾(黄箭头)。图9 双手X 线检查正位片示:双手各掌指骨及腕关节组成骨骨质密度轻度减低,以关节附近为甚,双侧尺桡骨远端及掌指骨骨骺未完全闭合。
讨论先天性肾上腺皮质增生症(Congenital adrenal hyperplasia,CAH)为常染色体隐性遗传病,其发病率为1/(10 000~20 000)[1]。17α-羟化酶缺乏症是CAH 的一种比较少见类型,约占1%,其发病与CYPl7A1 基因突变有关[2]。首例17α-羟化酶缺乏症是由Biglieri 等[3]在1966 年报道,迄今为止,全世界报道约200 余例。
CYP17A1 基因发生突变,17α-羟化酶部分或完全缺乏,导致肾上腺皮质类固醇激素合成障碍,从而引起皮质醇、雌二醇及睾酮合成障碍。当皮质醇缺乏时,激活下丘脑和垂体反馈调节系统,引起促肾上腺皮质激素(ACTH)分泌增加,从而刺激肾上腺皮质增生,同时由于孕酮堆积引起皮质酮及去氧皮质酮分泌增多,促使肾脏保钠排钾,钾流失出现低血钾及肌无力,血容量增加出现高血压。性激素缺乏时,女性患者(染色体核型46,XX) 由于苗勒管发育障碍出现内生殖器未发育或发育不良,原发性闭经,第二性征未发育;男性患者(染色体核型46,XY) 由于胚胎期睾酮不足不能形成阴茎及阴囊,而分化呈女性外生殖器,睾丸分泌的抗苗勒管激素使副中肾管退化,因此无子宫附件,性腺为发育不良的睾丸,从而形成男性假两性畸形。因此有学者认为以下为诊断17α-羟化酶缺乏症标准[4]:①顽固性高血压;②低血钾;③血皮质醇、睾酮和雌二醇水平低下,ACTH 升高;④原发性闭经,青春期第二性征未发育,女性内生殖器未发育或发育不良伴或不伴有隐睾;⑤肾上腺皮质增生;⑥骨质疏松;⑦骨龄延迟;⑧染色体核型为(46,XX)或(46,XY)。
肾上腺影像表现: 多层螺旋CT 薄层扫描为肾上腺首选影像检查方法,当患者皮质醇缺乏时,反馈性引起ACTH 分泌增多从而刺激肾上腺皮质增生,影像表现为单侧[1,5-6,8]或双侧肾上腺弥漫性或结节状增生肥大,有报道经糖皮质激素治疗后肾上腺结节可逐渐消失[7],也有肾上腺增生合并肿瘤报道[1]。本例患者为双侧肾上腺弥漫性增大伴多发等低混杂密度结节及肿块影,考虑肾上腺增生合并肿瘤可能。
生殖系统影像表现:女性胚胎(染色体核型46,XX)多表现为无子宫或幼稚子宫[9-10],卵巢未发育或发育不良(卵巢体积小或见单纯性囊肿)[10-11],部分卵巢可发育正常[9],无阴道或阴道呈长约3~9 cm 不等盲端[1,12-14];男性胚胎(染色体核型46,XY)表现女性外生殖器,无子宫及附件,无阴道或仅有阴道盲端,腹盆腔或腹股沟区可见软组织结节呈黄豆至花生米大的发育不良的隐睾[1,4,13-14]。本例患者染色体核型(46,XY),为女性外生殖器,无子宫及附件,阴道呈长约4 cm 盲端,双侧腹股区可见10 mm 大小发育不良的隐睾,完全符合17α-羟化酶缺乏症染色体核型(46,XY)生殖系统影像表现。
骨关节系统影像表现:骨关节系统检查方法首选X 线和CT,由于患者体内雄激素及雌激素合成障碍,表现为全身骨关节广泛性骨质密度轻度-重度减低,骨小梁稀疏,部分可合并有骨折[12],骨龄小于实际年龄[13],骨骺延迟闭合,从而导致成年后身高仍可持续增长[7]。本例患者双手及腕关节组成骨轻度骨质疏松,胸腰椎骨质疏松,27 岁骨骺仍未闭合。
17α-羟化酶缺乏症鉴别诊断: ①卵巢发育不全(Turner综合征),患者除了有高血压、原发性闭经、第二性征未发育或发育不良、子宫发育不良及骨质疏松等表现外,还具有高鄂弓、蹼颈、后发际低、肘外翻、脊柱畸形及身材矮小等Turner 特征[15]。②(46,XY)单纯性腺发育不全,两者共同点有原发闭经、女性外生殖器、女性内生殖器发育幼稚、青春期女性第二性征未发育或发育不良、骨质疏松等,(46,XY)单纯性腺发育不全患者还具有性腺为条索样组织,上肢较长、指距大于身高,骨龄与实际年龄基本符合,人工周期有撤退性出血,而17α-羟化酶缺乏症性腺为发育不良的睾丸,身高比例正常,骨龄小于实际年龄,人工周期无反应,常伴有高血压及低血钾[16]。
总之,当影像科医师发现患者有肾上腺增生,内生殖器未发育或发育不良,腹盆腔或双侧腹股沟区可见发育不良隐睾,骨质疏松及骨骺延迟闭合等影像学特征时,应当提示有17α-羟化酶缺乏症,建议临床医师行相关临床、实验室检查以及染色体核型检查确诊,以避免漏诊、误诊。
