氨基甲磺酸的高效简便合成
2019-06-13潘劲松王玉禄苏俊杰范玉明
潘劲松,王玉禄 ,苏俊杰,范玉明
(山东省工业和信息化研究院, 山东 济南 250013)
氨基甲磺酸具有优良的TNFα生成抑制效果,可直接作为肝脏疾病用药物使用[1]。其分子内含有一个氨基和一个磺酸基,具有较强的化学反应活性,可用于合成头孢尼西侧链、N-膦酰基甘氨酸、以及抗纤维蛋白溶素剂等[2-4]。此外,还具有其他氨基磺酸类产品通用性能,可广泛用于日化清洗、固体催化、酸性蚀刻、离子液体脱硫脱碳等领域。
氨基甲磺酸从1905年被首次合成以来,其合成工艺路线主要有以下两条:
工艺路线一是以亚硫酸铵、甲醛为起始原料,经反应生成氨基甲烷磺酸铵,再酸化处理得到氨基甲磺酸[5。

此路线,中间体氨基甲磺酸铵容易与甲醛发生缩合反应,目标产物收率极低,提纯非常困难。
工艺路线二是以亚硫酸钠、甲醛为起始原料,经反应生成羟基甲烷磺酸钠,羟基甲烷磺酸钠与氨水进行氨解反应得到氨基甲烷磺酸钠后,酸化处理得到氨基甲磺酸[6-8]。
此路线,中间产物羟基甲烷磺酸钠在用氨水氨解时容易发生深度氨解,造成产物中伯胺类化合物选择性较低。此外,此工艺副产大量硫酸钠,给产品的分离与精制带来较大麻烦。
本文以甲醛、氨水、气体二氧化硫为原料,采用"一锅法"直接合成氨基甲磺酸。
与上述两条路线相比,直接采用二氧化硫进行磺化,可有效控制副反应发生,避免氯化铵、硫酸钠等无机盐副产物产生,所得产品容易分离与精制。
1 合成实验
1.1 采用二氧化硫制备氨基甲磺酸实验
按照图1,组装好实验装置。
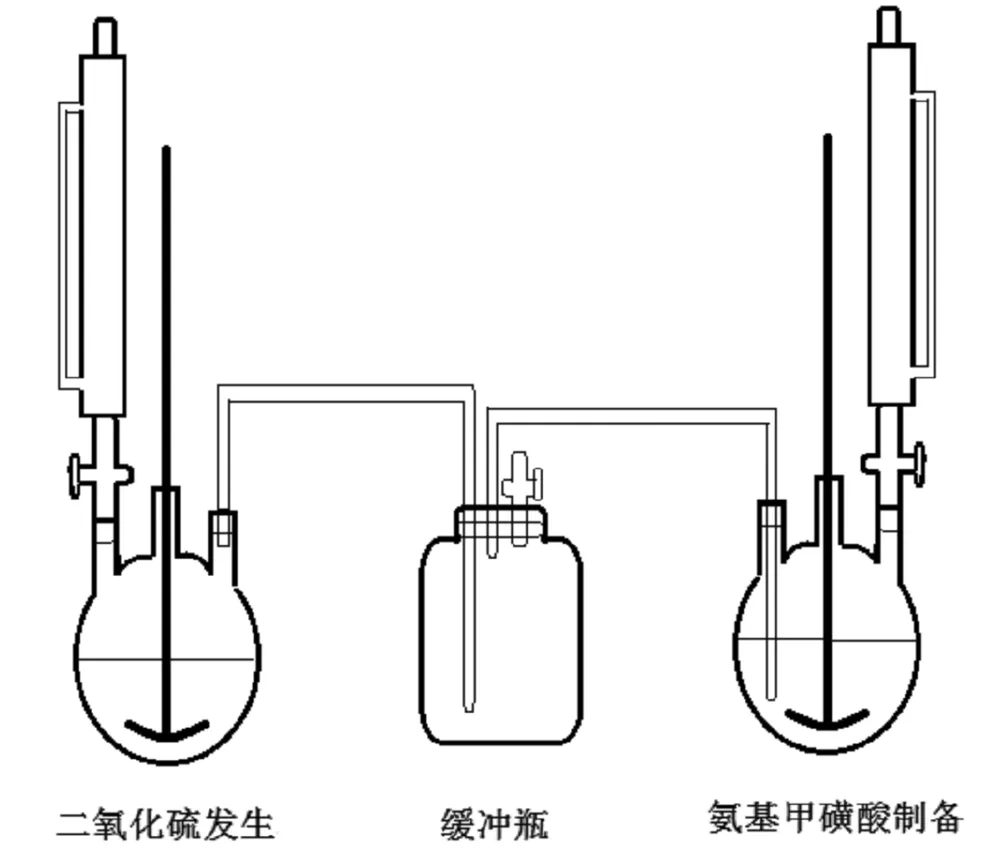
图1 三氧化硫制备氨基甲磺酸实验装置图
将氨基甲磺酸制备三口烧瓶置于冰浴上,向三口烧瓶内加入152mL甲醛(36.5%),向滴液漏斗内加入137mL氨水(27.5%)。开启搅拌,控制内温低于10℃以下,然后缓慢滴加氨水。滴加期间,控制三口烧瓶内温≤20℃。滴毕,保温反应2h后,再升温至30℃,继续反应2h。
检查二氧化硫装置气密性,确保二氧化硫完全进入氨基甲磺酸制备烧瓶内。向二氧化硫三口烧瓶内加入458g亚硫酸氢钠、1500mL水,开启搅拌,至亚硫酸氢钠全部溶解,向恒压漏斗内加入125mL浓硫酸(98%)。关闭缓冲瓶放空阀。
在氨基甲磺酸制备烧瓶内温降至10℃以下后,开始缓慢滴加浓硫酸,控制二氧化硫发生烧瓶内温60~80℃。同时,控制氨基甲磺酸制备烧瓶内温≤15℃。通入二氧化硫结束后,继续维持内温20±2℃,反应4h。然后将反应瓶降温至0℃,过滤,滤饼用冰水洗涤,晾干,得到白色固体190g(记作样品A),产品收率85.5%。
1.2 采用亚硫酸钠直接制备氨基甲磺酸对比实验
向1000mL三口烧瓶内加入230g亚硫酸氢钠、250mL水,开启搅拌,至亚硫酸氢钠全部溶解。向滴液漏斗内加入152mL甲醛(36.5%),缓慢升温至70℃,保温反应4h。然后滴加137mL氨水(27.5%),70℃下反应2h。再降温至25℃以下,缓慢滴加硫酸,中和至pH值<2。继续降温至0℃,保温2h,然后过滤,所得产品用蒸馏水进行重结晶、洗涤,晾干,得到白色固体55g(记作样品B),产品收率24.7%。
2 产品分析
2.1 熔点
采用YRT-3型熔点测定仪,检测样品A和样品B熔点。
样品A:达到183℃后,试样迅速分解。与标准品分解温度184℃(文献值),基本一致。
样品B:达到180℃以上,试样部分分解,继续升温至200℃以上,仍有部分不熔。分析试样可能含有无机盐或者其他副产物。
2.2 含量
采用电位滴定法,用氨基甲磺酸标准品确定滴定曲线的等当点后,测定样品A和样品B含量。
样品A:含量99.2%,与进口试剂标准品相当。
样品B:含量65.5%。分析试样可能含有其他杂质。
2.3 延伸实验
参照文献[9]投料比及工艺条件,分别以50g样品A和样品B为起始原料合成头孢尼西侧链。将氨基甲磺酸样品与氢氧化钾反应,得到氨基甲磺酸钾后,再将氨基甲磺酸钾在碱性条件下,与二硫化碳反应得到磺甲基二硫代氨基甲酸钾。磺甲基二硫代氨基甲酸钾与溴乙烷经烷基化反应,得到磺甲基二硫代氨基甲酸乙酯的钾盐后,再与叠氮化钠进行环合反应。环合反应产物再经离子交换、成盐等步骤,得到头孢尼西侧链产物。合成路线如下。
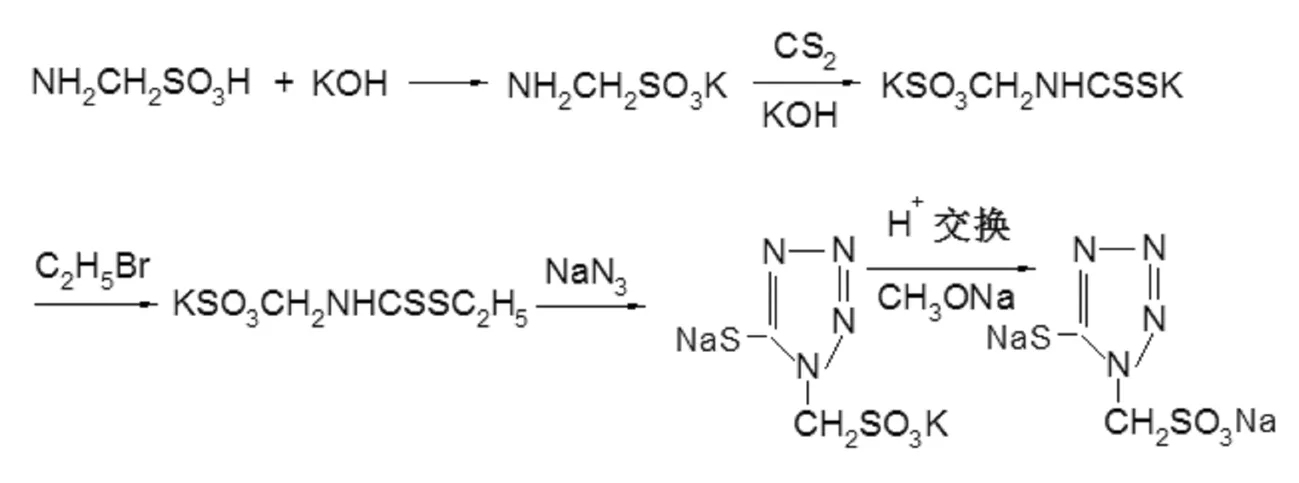
样品A:投料比、工艺参数等与参考文献一致,最终得到头孢尼西侧链产品15g。经HPLC测定,其纯度为98.5%。
样品B:投料比、工艺参数与样品A保持一致,但最终未能得到固体产品。分析可能由于试样纯度低或试样杂质造成其他副反应,直接影响了目标产物的生成。
3 结论
通过以上实验对比,直接采用二氧化硫合成氨基甲磺酸新工艺,不仅能得到符合下游合成要求的合格产品,而且在产品收率、产品提纯等方面都优于传统工艺。该工艺路线过程简单、效率高,具有很高的工业化应用价值。此外,此工艺路线也给脱除烟气中SO2提供了一种思路借鉴,为综合利用烟气中SO2提供了一种方法。
