儿童朗格汉斯细胞组织细胞增生症的诊治新进展
2019-05-24龙庆玲李长钢
龙庆玲,李长钢,2
朗格汉斯细胞组织细胞增生症(Langerhans cell histiocytosis,LCH)是血液系统的一种罕见病,是一组以CD1a+、CD207+树突状细胞异常增生为特点的组织细胞增生性疾病,临床表现复杂多样,可侵犯单器官、多器官及多系统,以骨骼、皮肤及垂体损伤常见。目前发病机制尚不清楚,且缺乏有效、快速的诊断手段,因此,笔者于2017年12月对近年来的最新研究进行收集整理,现综述如下。
1 定义及流行病特征
组织细胞起源于骨髓干细胞,包括单核巨噬细胞(monocyte/macrophages,MC)和树突状细胞(dendritic cell,DC),成熟DC表面可特征性表达CD1a、CD11c、CD83等,而朗格汉斯细胞(Langerhans cell,LC)是特异性表达CD207+、CD1a+的表皮DC[1],有抗原呈递及调节免疫的作用[2]。组织细胞增多症是以组织细胞异常增生为共同特点的一组疾病,分为LCH、非LCH及肿瘤性。LCH是一组以CD1a+、CD207+DC异常增生为特点的组织细胞增生性疾病[3],可侵犯全身各器官及系统,以骨骼、皮肤、垂体损伤常见。该病临床罕见,好发于儿童,儿童发病率为5/10万[4],肺LCH(pulmonary Langerhans cell histiocytosis,PLCH)男女发病率分别为2.7/100万、0.7/100万,也有家族病例及青少年LCH经治疗后并发白血病等个案报道[5],目前尚无地方发病的统计。
2 LCH发病机制
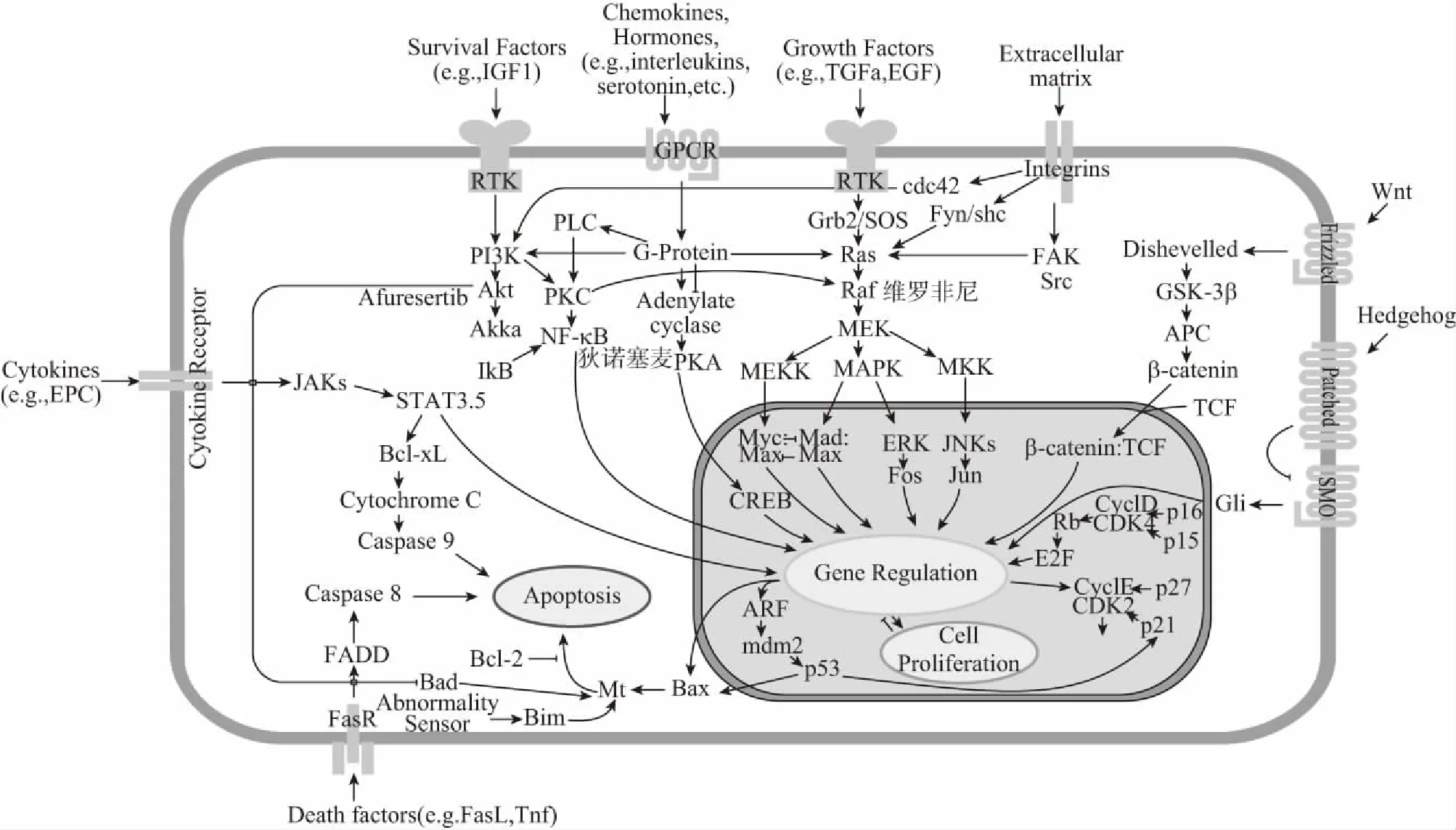
注:羟基脲主要能抑制DNA合成,从而抑制朗格汉斯细胞的导常增生;来那度胺与地塞米松均能有效抑制炎症反应,有效治疗LCH图1 RAF/RAS/MEK信号通路及各靶向药物作用靶点图
LCH致病机制尚不明确,目前主要有炎症反应性假说和肿瘤增生性假说;丝/苏氨酸蛋白激酶基因——BRAF基因突变在LCH中的发现使其发病机制取得了突破性进展,在骨髓干细胞发现BRAFV600E基因突变、LCH髓系前体细胞表达BRAFV600E突变等均强烈支持该病为髓源性组织细胞肿瘤样增生性疾病[6],因此更倾向于肿瘤增生性病变。BRAF基因又称B-Raf原癌基因,主要编码B-Raf蛋白,现已有30多种突变体。研究发现50%以上LCH存在BARF突变,其中38%~64%为BRAFV600E突变,而BRAFV600E突变能激活RAS/RAF/MEK通路,使丝裂原激活蛋白酶(mitogen-activated protein kinase,MAPK)持续活化,活化核因子κb(nuclear factor kappa-B,NF-κb),最终引起前炎性因子白细胞介素-1(interleukin-1,IL-1)、白细胞介素-6(interleukin-6,IL-6)、肿瘤坏死因子-α(tumor necrosis factor-α,TNF-α)等大量释放(如图1所示);同时还能延缓瘤细胞凋亡,增加肿瘤的发病;目前该通路还发现MAP2K1、ARAF、ERBB3、PIK3CA等多种基因突变[7]。另有研究发现PLCH的发病与吸烟密切相关,主要与吸烟能使CCR6、CCR7发生调节变异相关,但具体致病机制尚未明确[2]。
病理学研究发现,不同类型LCH的生物学行为也存在一定差异,单系统LCH(single system LCH,SS-LCH)多以延长DC寿命为特点的良性增生,预后良好;但多系统LHC(multi-system LCH,MS-LCH)则为侵袭性生长,SS-LCH、高危多系统(risk organ multi-system LCH,RO-MS-LCH)、低危多系统(low-risk organ multi-system LCH,LRO-MS-LCH)可迅速侵犯多个器官及多个系统,甚至迅速侵犯高危器官(如血液、肝脾等)而危及生命,预后差,复发及后遗症发生率高[8]。
3 临床表现及分类
临床症状根据侵犯部位及范围不同而轻重不一,以骨骼、皮肤及垂体损伤常见,还包括肺、淋巴结及中枢等。LCH以单灶性骨损伤最常见,但2岁内儿童则以多灶性骨损伤为主,依次为颅骨、股骨、面颅骨、椎骨及盆骨等损伤,表现为疼痛、跛行、病理性骨折、红肿等;侵犯皮肤可见斑丘疹、湿疹及合并脓肿、结痂的特异性皮疹,40%累及多系统;垂体受侵则出现多尿、生长激素缺乏症;血液系统受侵出现面色苍白、出血或感染等;肺累及时出现气促、呼吸窘迫等;肝脏以肝功能障碍为主。LCH可分成SS-LCH、RO-MS-LCH、LRO-MS-LCH及特定部位LCH四类。SS-LCH最常见,是指仅侵犯单个组织、器官或系统,25%可进展为MS-LCH,有研究发现HLA-DEB18*03+的淋巴细胞能阻止其向MS-LCH发展。MS-LCH多见于婴幼儿,是指累及两个或两个以上器官或系统。该病约30%~40%可留下后遗症[9-10]。PLCH是一种罕见的肺间质疾病,以40岁以下吸烟者多见,主要特点以Langerin+DCs在肺间质沉积为特点,形成细支气管中心结节及肺囊性重构,此类病人多死于呼吸衰竭等严重并发症。神经组织退化性LCH(neurodegenerative langerhans cell histiocytosis,ND-LCH) 常见于中枢受损后10年,包括构音障碍、共济失调、排尿障碍和行为改变等,儿童以迟发性精神运动障碍最常见,预后差[6]。
4 诊断新进展
4.1 病理诊断现在病理诊断仍为LCH的金标准[11-12];近年发现CD207+即Langerin特异性表达于Birbeck颗粒表面,因此,CD207+可取代Birbeck颗粒成为新的诊断标准之一。另外,S100蛋白在LCH中也显著升高,Ki-67则与预后密切相关[13]。
4.2 液体活检2013年Haupt等[14]在实体瘤血液中检出BRAFV600E的循环游离DNA(circulating cell-free DNA,ccfDNA),被称为液体活检,未来可能会取代病理活检成为确诊的新技术,并能快速诊断伴高风险活检的病变。另外,BRAFV600E的ccfDNA定量检测将可能成为监测病情的标志物[15]。
5 治疗及预后
5.1 传统治疗根据2013年文献[14]研究所示,治疗前评估对其疗效、预后起着重要作用。危险器官是指侵犯某些器官并死于该器官病变的并发症,如血液、淋巴系统、肝脾等;在LCH的四期临床研究(LCH-Ⅳ)中,肺将不再作为高危器官,中枢神经系统危险病变指侵犯某些器官是发生神经退性损伤的标志,如头颅骨损伤可预测尿崩症(DI)和中枢神经系统病变或ND-LCH。目前无生物标志物可评估活动性,未来将重点研究ccfDNA定量检测。
孤立性骨骼或皮肤损伤均有一定自限性,小病灶骨损伤(<5 cm)可部分或完全刮除,孤立性皮肤或淋巴结病变可给予手术切除、局部激素、氮芥等治疗;除上述情况外,其他均应系统治疗。DI损伤不可逆,因此DI不是系统治疗的指针,活动期才需系统治疗。PLCH易发生严重并发症,常予低量激素、克拉屈滨等联合治疗[16]。标准/一线方案以长春碱(VBL)和激素为基础,分强化诱导、维持治疗两个阶段:(1)强化诱导,VBL 静脉注射,每次6 mg/m2, 1次/周,共6周;泼尼松 40 mg·m-2·d-1,3次/天,共4周,后2周逐渐减量(共1~2个周期);(2)维持治疗,VBL静脉注射,每次6 mg/m2,1次/3周;泼尼松 40 mg·m-2·d-1,3次/天,每周给药5 d,每3周一个循环,疗程12个月可减少复发,但继续延长维持治疗疗程是否进一步减少复发还有待更为深入的研究;(3)进行一个周期强化治疗后仍有活动病灶时,可再次强化诱导,RO-MS-LCH可加用巯嘌呤 50 mg·m-2·d-1。完成诱导化疗及疾病治疗过程中,应及时进行疗效评价,即(1)好转:完全缓解或疾病完全消退;(2)不变:原病灶不变,而出现新的病灶;(3)恶化:疾病进展。
5.2 二线方案对于一线治疗失败、复发及难治性LCH,应积极行二线治疗。可选择化疗联合克拉屈滨及阿糖胞苷。有报道阿糖胞苷联合克拉屈滨治疗RO-MS-LCH,有效率92%,5年生存率82%;若病变主要侵犯非危险器官,可单用克拉屈滨或联合VBL和类固醇治疗[17]。目前针对MAPK通路的靶向药物已成为研究热点,维罗非尼作为BRAF抑制剂,对BRAFV600E突变者疗效显著;有报道其能治愈RO-MS-LCH[18],但临床研究发现20%~50%成人可发生原发性鳞状细胞癌,因此,还不能推荐为一线药物;但对于伴发中枢或心血管等严重并发症或一线治疗无效的病人可首选[19-20]。狄诺塞麦是NF-κb受体激活物配体的抑制剂,能阻断前炎性因子释放,有效减轻炎症反应,已有该药已成功治愈2例成人LCH的报道[21]。Afuresertib是一种有效的AKT抑制剂,能有效抑制细胞生长和增殖,在LCH中已进入Ⅱ期临床研究[22]。而上述分子靶向药物费用昂贵,在经济贫困地区应用有效。因此,有研究显示用地塞米松+来那度胺成功治愈了4例复发性儿童LCH,随访15~18个月均达到缓解,尚无不良事件[3,23],从而得出一个有效、安全、廉价及可行性强的新方案:地塞米松(第1、8、15、21天给予0.8 mg/kg)、来那度胺(体质量≤15 kg者2.5 mg/d,体质量>15 kg者第1~21天5.0 mg/d),每个疗程28 d,共6个疗程[24]。羟基脲(HU)用于LCH是老药新用,能抑制DNA合成,发现单用HU或联合甲氨蝶呤治疗复发或难治性LCH,80%部分或完全缓解,无严重毒不良反应,是一种安全有效、经济可行的新方案[25-26]。
另外,多数专家认为放疗能诱发肿瘤、影响儿童生长发育等毒副反应显著,而不推荐在儿童中使用,但也有专家认为对青少年孤立性椎体损伤进行放疗能有效缩小病灶、缓解疼痛及减少并发症等,儿童剂量为2.5~45.0 Gy,推荐累计剂量为<7.5 Gy。最后,器官移植长期疗效目前尚不明确,可作为上述治疗失败后的尝试手段,但有研究报道移植前后去甲基化治疗能增加其疗效,减少并发症[10]。
5.3 治疗新进展传统的二线方案费用昂贵,在经济欠发达地区应用有限。因此,学者们[3,27]用地塞米松+来那度胺成功治愈4例复发性儿童LCH,随访15~18个月均达到缓解,尚无不良事件,从而得出一个有效、安全、廉价及可行性强的新方案:地塞米松+来那度胺[23]。目前针对MAPK通路的靶向药物已成为研究的热点,维罗非尼等是BRAF的抑制剂,对BRAFV600E突变疗效显著,以有成功治愈的报道。
5.4 预后LCH总生存率较高,SS-LCH高达100%,伴或不伴高危器官的MS-LCH,5年生存率分别为87%、100%,而5年复发率分别为25%和37%,儿童LCH超过1/3并发后遗症,包括骨骼畸形、内分泌功能障碍、听力障碍、神经及心理问题等,长期预后差[27]。Maia等[7]报道儿童与成人总生存率差异无统计学意义,且成人的复发及病死率更高。BRAFV600E常见于青少年,BRAFV600E突变的儿童LCH病情危重、一线方案疗效差、后遗症发生率显著升高。另外,ND-LCH全身化疗不能减少DI的发生,但其复发和活动期延长却能增加发病。因此,长期随访对其预后至关重要[23]。
综上所述,LCH是一组以树突状细胞异常增生为特点的组织细胞增生性疾病,临床常侵犯单器官、多器官及多系统,以骨骼、皮肤和垂体多见。其发病机制目前更倾向于肿瘤增生性假说,主要与MAPK通路异常活化相关,但具体作用机制有待进一步研究。病理仍为诊断的金标准,但CD207+检测可替代电镜Birbeck颗粒检查成为新的诊断标准;更重要的是,液体活检技术将有望取代传统的组织活检,成为新的无创性确诊手段,而ccfDNA的定量检测则可能成为病情监测的首个生物标志物。标准化疗仍为一线方案,但近年来已将研究的热点转向分子靶向药物并将可能取代传统化疗,成为新的首选治疗方案;未来应积极进行临床实验研究,为儿童安全、有效的应用提供更多依据。其次,新方案(地塞米松+来那度胺)在贫困地区将有望取代昂贵的新型靶向药物,成为首选的二线方案;此外,HU用于LCH是老药新用,疗效显著,未来将重点开展HU与MTX、巯嘌呤及观察组间的前瞻性研究。虽然器官移植的长期不良事件明显,但移植前后进行去甲基化治疗可望减少其不良事件的发生,值得更深层次的研究。
